A Numpy-based PDB structure manipulation package

Project description
Afpdb - An Efficient Protein Structure Manipulation Tool


The advent of AlphaFold and other protein AI models has transformed protein design, necessitating efficient handling of large-scale data and complex workflows. Traditional programming packages, developed before these AI advancements, often lead to inefficiencies in coding and slow execution. To bridge this gap, we introduce Afpdb, a high-performance Python module built on AlphaFold’s NumPy architecture. Afpdb leverages RFDiffusion's contig syntax to streamline residue and atom selection, making coding simpler and more readable. By integrating PyMOL’s visualization capabilities, Afpdb enables automatic visual quality control, enhancing productivity in structural biology. With over 190 methods commonly used in protein AI design, Afpdb supports the development of concise, high-performance code, addressing the limitations of existing tools like Biopython. Afpdb is designed to complement powerful AI models such as AlphaFold and ProteinMPNN, providing the additional utility needed to effectively manipulate protein structures and drive innovation in protein design.
Automatic chain alignment, antibody analysis, simplified PyMOL visualization for multiple protein objects, and many methods have been added in version 0.3. The algorithm to identify gaps within a chain are improved to minimize false gaps. Tests, docstring, and argument typing are improved to help Github Coplit to understand the module better. To explore these new features, please search for the tag "#2025JUL" in the Notebook or tutorial documentation.
Please read our short artical published in Bioinformatics.

Tutorial
The tutorial book is availabe in PDF.
The best way to learn and practice Afpdb is to open Tutorial Notebook in Google Colab.
Table of Content
- Demo
- Fundamental Concepts
- Internal Data Structure
- Contig
- Selection
- Atom Selection
- Residue Selection
- Residue List
- Read/Write
- PDB Information
- Sequence & Chain
- Extarction
- Missing Residues
- REsidue Numbering
- Geometry, Measurement, & Visualization
- Select Neighboring Residues
- Display
- B-factors
- PyMOL Interface
- PyMOL3D Display
- RMSD
- Solvent-Accessible Surface Area (SASA)
- Secondary Structures - DSSP
- Bond Length
- Internal Coordinates
- Object Manipulation
- Move Objects
- Align
- Automatic Chain Mapping & Alignment
- Split & Merge Objects
- Antibodies
- CDR Identification
- CDR Visualization
- scFv Analysis
- Parsers for AI Models
AI Use Cases
Interested in applying Afpdb to AI protein design? Open AI Use Case Notebook in Google Colab.
Table of Content
- Example AI Protein Design Use Cases
- Handle Missing Residues in AlphaFold Prediction
- Structure Prediction with ESMFold
- Create Side Chains for de novo Designed Proteins
- Compute Binding Scores in EvoPro
Developer's Note
Open Developer Notebook in Google Colab.
Install
Stable version:
pip install afpdb
or
conda install bioconda::afpdb
Development version:
pip install git+https://github.com/data2code/afpdb.git
or
git clone https://github.com/data2code/afpdb.git
cd afpdb
pip install .
To import the package use:
from afpdb.afpdb import Protein,RS,RL,ATS
Demo
Structure Read & Summary
# load the ab-ag complex structure 5CIL from PDB
p=Protein("5cil")
# show key statistics summary of the structure
p.summary().display()
Output
Chain Sequence Length #Missing Residues #Insertion Code First Residue Name Last Residue Name
-- ------- ---------------------------------------------------------------------------------------------------------------------
0 H VQLVQSGAEVKRPGSSVTVS... 220 8 14 2 227
1 L EIVLTQSPGTQSLSPGERAT... 212 0 1 1 211
2 P NWFDITNWLWYIK 13 0 0 671 683
Residue Relabeling
print("Old P chain residue numbering:", p.rs("P").name(), "\n")
Output:
Old P chain residue numbering: ['671', '672', '673', '674', '675', '676', '677', '678', '679', '680', '681', '682', '683']
p.renumber("RESTART", inplace=True)
print("New P chain residue numbering:", p.rs("P").name(), "\n")
Output:
New P chain residue numbering: ['1', '2', '3', '4', '5', '6', '7', '8', '9', '10', '11', '12', '13']
p.summary()
Output
Chain Sequence Length #Missing Residues #Insertion Code First Residue Name Last Residue Name
-- ------- ---------------------------------------------------------------------------------------------------------------------
0 H VQLVQSGAEVKRPGSSVTVS... 220 8 14 1 226
1 L EIVLTQSPGTQSLSPGERAT... 212 0 1 1 211
2 P NWFDITNWLWYIK 13 0 0 1 13
Replace Missing Residues for AI Prediction
print("Sequence for AlphaFold modeling, with missing residues replaced by Glycine:")
print(">5cil\n"+p.seq(gap="G")+"\n")
Output
Sequence for AlphaFold modeling, with missing residues replaced by Glycine:
>5cil
VQLVQSGAEVKRPGSSVTVSCKASGGSFSTYALSWVRQAPGRGLEWMGGVIPLLTITNYAPRFQGRITITADRSTSTAYLELNSLRPEDTAVYYCAREGTTGDGDLGKPIGAFAHWGQGTLVTVSSASTKGPSVFPLAPSGGGGGGGGGTAALGCLVKDYFPEPVTVGSWGGGGNSGALTSGGVHTFPAVLQSGSGLYSLSSVVTVPSSSLGTGGQGTYICNVNHKPSNTKVDKKGGVEP:EIVLTQSPGTQSLSPGERATLSCRASQSVGNNKLAWYQQRPGQAPRLLIYGASSRPSGVADRFSGSGSGTDFTLTISRLEPEDFAVYYCQQYGQSLSTFGQGTKVEVKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNR:NWFDITNWLWYIK
Interface Computing
# identify H,L chain residues within 4A to antigen P chain
rs_binder, rs_seed, df_dist=p.rs_around("P", dist=4)
# show the distance of binder residues to antigen P chain
df_dist[:5].display()
Output
chain_a resn_a resn_i_a resi_a res_a chain_b resn_b resn_i_b resi_b res_b dist atom_a atom_b
--- --------- -------- ---------- -------- ------- --------- -------- ---------- -------- ------- ------- -------- --------
408 P 6 6 437 T H 94 94 97 E 2.63625 OG1 OE2
640 P 4 4 435 D L 32 32 252 K 2.81482 OD1 NZ
807 P 2 2 433 W L 94 94 314 S 2.91194 N OG
767 P 1 1 432 N L 91 91 311 Y 2.9295 ND2 O
526 P 7 7 438 N H 99E 99 107 K 3.03857 ND2 CE
Residue Selection & Boolean Operations
# create a new PDB file only containing the antigen and binder residues
q=p.extract(rs_binder | "P")
Structure I/O
# save the new structure into a local PDB file
q.save("binders.pdb")
Structure Display within Jupyter Notebook
# display the PDB struture, default is show ribbon and color by chains.
q.show(show_sidechains=True)
Output (It will be 3D interactive within Jupyter Notebook)
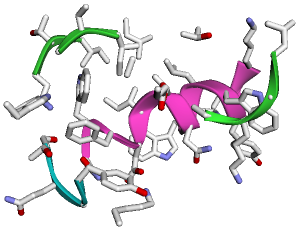
Antibody Analysis & PyMOL Visualization
# identify the variable domain, CDR regions, use B-factors to represent different CDR regions
rs_cdr, rs_var, c_chain_type, c_cdr = p.rs_antibody(scheme="imgt", set_b_factor=True)
# remove constant domain, only keep the variable domain for antibody
# we translate the coordindate of the truncated protein by a small amount to avoid q being shadowed by p in the display
q=p.extract(rs_var | p.rs("P")).translate([0.5, 0.5, 0.5], inplace=True)
print(q.len_dict())
# The truncated antibody is smaller, as it only contains the variable domain
# color the full antibody variable+constant in white
# The truncated variable-only object in rainbow color
Protein.PyMOL3D() \
.show(p, color="white") \
.show(q, color="spectrum") \
.show(output="myAb.pse", save_png=True, width=250, height=250)
# A PyMOL session file and png file are generated.
Output (myAb.pse can be opened with PyMOL)
{'H': 220, 'L': 212, 'P': 13}
{'H': 126, 'L': 108, 'P': 13}
Save: Please wait -- writing session file...
Save: wrote "myAb.pse".
PyMOL session saved: myAb.pse
High-quality image saved: myAb.png
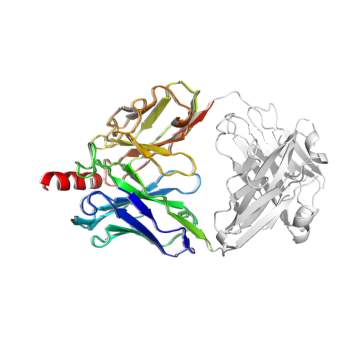
Automatic Object Alignment
# We remove constant domain, change chain names, and translate the new protein
q = p.truncate_antibody() \
.rename_chains({"H":"X", "L":"Y", "P":"Z"}) \
.translate([3, 4, 5], inplace=True)
# The new protein is shorter
print(q.len_dict())
# Automatic chain pairing and sequence alignment
p2, q2, rl_p, rl_q = p.align_two(q, auto_chain_map=True)
# aligned portion
# The alignment output indicates it figures out chain H/L/P should be paired with X/Y/Z.
print("Original protein p:", rl_p, "\n")
print("Truncated protein q:", rl_q, "\n")
print("RMSD (expecting 0):", p.rmsd(q, rl_p, rl_q, align=True))
Output
{'X': 126, 'Y': 108, 'Z': 13}
Chain Mapped: H <> X
target 0 VQLVQSGAEVKRPGSSVTVSCKASGGSFSTYALSWVRQAPGRGLEWMGGVIPLLTITNYA
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
query 0 VQLVQSGAEVKRPGSSVTVSCKASGGSFSTYALSWVRQAPGRGLEWMGGVIPLLTITNYA
target 60 PRFQGRITITADRSTSTAYLELNSLRPEDTAVYYCAREGTTGDGDLGKPIGAFAHWGQGT
60 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
query 60 PRFQGRITITADRSTSTAYLELNSLRPEDTAVYYCAREGTTGDGDLGKPIGAFAHWGQGT
target 120 LVTVSS 126
120 |||||| 126
query 120 LVTVSS 126
Chain Mapped: L <> Y
target 0 EIVLTQSPGTQSLSPGERATLSCRASQSVGNNKLAWYQQRPGQAPRLLIYGASSRPSGVA
0 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
query 0 EIVLTQSPGTQSLSPGERATLSCRASQSVGNNKLAWYQQRPGQAPRLLIYGASSRPSGVA
target 60 DRFSGSGSGTDFTLTISRLEPEDFAVYYCQQYGQSLSTFGQGTKVEVK 108
60 |||||||||||||||||||||||||||||||||||||||||||||||| 108
query 60 DRFSGSGSGTDFTLTISRLEPEDFAVYYCQQYGQSLSTFGQGTKVEVK 108
Chain Mapped: P <> Z
target 0 NWFDITNWLWYIK 13
0 ||||||||||||| 13
query 0 NWFDITNWLWYIK 13
Original protein p: H2-113:L1-107:P
Truncated protein q: X:Y:Z
RMSD (expecting 0): 7.16936768611062e-15


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file afpdb-0.3.1.tar.gz.
File metadata
- Download URL: afpdb-0.3.1.tar.gz
- Upload date:
- Size: 139.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.9.15
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
bd2b3cf09401a305be09cb8f8518bd83343f94e6969b6b2f5fd5cb8c0da32b4b
|
|
| MD5 |
5ef2dc7ef9c6fc19163e54e32161d3b7
|
|
| BLAKE2b-256 |
7722b6fa6a1ebde7660fb8e5513c5fce4949aa3c0701f3afab7b38770f5c15c5
|
File details
Details for the file afpdb-0.3.1-py3-none-any.whl.
File metadata
- Download URL: afpdb-0.3.1-py3-none-any.whl
- Upload date:
- Size: 128.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.9.15
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a2593a7b750e4839c1d5919ac46e416d99201f4b1240863ae3babe7be54d550d
|
|
| MD5 |
053c5e7abd10c15cd34838c534ee3d4b
|
|
| BLAKE2b-256 |
5c2b1e49d4cc27d9a00ec472bd2350b95ea7299b712589e96e10c339dff9d494
|







