a quality control tool for bacterial ONT-only assemblies

Project description
Alpaqa: Assembly-Level Profiling And Quality Assessment

Alpaqa analyzes base-level quality scores in bacterial genome assemblies generated from Oxford Nanopore sequencing data.
The tool masks noisy regions and quantifies the number of Low-Quality Bases (LQBs) per megabase, a metric we found to correlate with overall base-level assembly accuracy. In addition, Alpaqa performs binomial tests to identify 4-, 5-, and 6-mer motifs that are significantly associated with LQBs. These motifs often represent systematic, error-prone sequence contexts and may correspond to targets of DNA modification systems.
Alpaqa operates on polished assemblies generated with Dorado polish or Medaka2. By detecting systematic error signatures, Alpaqa allows users to flag assemblies that may appear complete but contain motif-specific inaccuracies. This is particularly valuable in applications requiring high base-level accuracy, such as outbreak investigation and transmission analysis.
An automated bacterial genome assembly pipeline for ONT data which includes Medaka2 polishing, quality assessment with alpaqa, and masking of low-quality bases is available at boap.
Installation
Via pip
Alpaqa can be installed directly from PyPI:
pip install alpaqa-bio
Via uv (recommended)
If you use uv, you can run it without installation:
uvx --from alpaqa-bio alpaqa --help
Or install it:
uv tool install alpaqa-bio
Generating input files for alpaqa
Alpaqa relies on fastq assembly files with phred quality scores generated with dorado polish or medaka2 (-q flag). The tool has been tested with data generated with SUP@v5.0, SUP@v5.2, HAC@v5.0, and HAC@v5.2 basecalling models. SUP data is strongly recommended.
Using medaka2
READS=reads.fastq.gz
ASSEMBLY=draft_assembly.fasta
THREADS=16
# medaka2
medaka_consensus -i $READS -d $ASSEMBLY -o polished_assembly -t $THREADS --bacteria -q
Using dorado polish
READS=reads.fastq.gz
ASSEMBLY=draft_assembly.fasta
THREADS=16
dorado aligner $ASSEMBLY $READS | samtools sort -@ $THREADS -o aligned.bam
samtools index aligned.bam
dorado polish aligned.bam $ASSEMBLY -t $THREADS -o polished_assembly --bacteria -q
Alpaqa usage
Once installed, run alpaqa by passing your FASTQ assembly files:
alpaqa -i *.fastq --threads 16
Options
usage: alpaqa -i assembly.fastq -o output.tsv --threads 16 [options]
ALPAQA v0.1.3
options:
-h, --help show this help message and exit
-i INPUT [INPUT ...], --input INPUT [INPUT ...]
Input fastq assembly file(s).
-o OUTPUT, --output OUTPUT
Output TSV filename. (default: alpaqa_report.tsv)
--report Generate detailed reports on kmer analysis.
-t THREADS, --threads THREADS
Number of threads. (default: 1)
-v, --version show program's version number and exit
Advanced:
--all-contigs Analyze ALL contigs > min_contig_len (Default is longest contig only).
--no-mask Disable masking of LQB dense regions.
--lqb-threshold LQB_THRESHOLD
Phred Q-score threshold (inclusive). (default: 5)
--min-contig-len MIN_CONTIG_LEN
Minimum contig length to include. (default: 50000)
--window-size WINDOW_SIZE
Window size (bp) for scanning LQB dense regions. (default: 5000)
--density-multiplier DENSITY_MULTIPLIER
Multiplier for baseline density to trigger masking. (default: 5.0)
--mask-floor MASK_FLOOR
Absolute minimum LQB density (0.001 = 0.1%). (default: 0.001)
Output
Alpaqa generates a tab-separated (TSV) report. Each row represents one input assembly file with the following fields:
| Field | Description |
|---|---|
| Filename | Name of the input FASTQ file. |
| AvgQ | Average Phred quality score across all bases in the file. |
| LQB_raw/Mbp | Low-Quality Bases per Megabase (Raw): Total count of bases with a Phred score between 1 and 5 in the entire file, normalized per million bases. |
| LQB/Mbp | Low-Quality Bases per Megabase (Filtered): The density of low-quality bases in the longst contig after excluding (masking) unreliable LQB-dense regions. |
| MaskThresh | Dynamic Masking Threshold: The density threshold that triggered masking. Calculated as 5x the baseline density, but is at least 0.1% (0.001) to prevent over-masking assemblies. |
| Masked_Bases | Total number of bases removed from the analysis. |
| Contigs_Analyzed | Shown as X//Y, where X is the number of contigs analyzed (default: longest contig only) and Y is the total number of contigs in the file. |
| Bases_Analyzed | Total number of bases analyzed. |
| Sig4m / 5m / 6m | Significant Motifs: The top 3 DNA patterns (4, 5, and 6-mers) most significantly linked to quality drops (Q1-5) based on binomial testing. Includes the motif and the percentage of its occurrences that were low quality. |
Interpretation
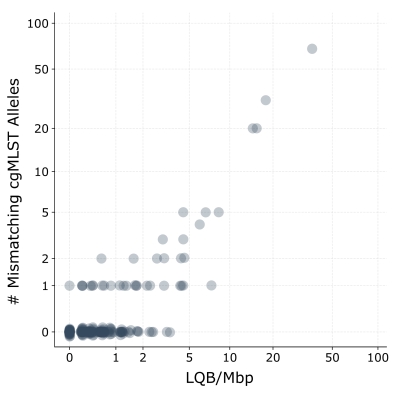
For assemblies generated with SUP@v5.2 data, following thresholds were established:
- <5 LQBs/Mbp: Highly accurate. These assemblies typically yield identical or near-identical cgMLST profiles when compared to Illumina-polished references.
- 5 to 10 LQBs/Mbp: Potentially reliable but should be interpreted with caution. Masking low-quality bases with fastq2a.py (see below) is recommended for downstream cgMLST analyses.
- >10 LQBs/Mbp: Usually unsuitable for high-resolution genotyping due to excessive base-level errors.
Beyond systematic sequencing errors, elevated LQB counts may indicate sample contamination, insufficient sequencing depth, or low read quality, which can be further investigated using tools such as nanoq and CheckM.
Masking low-quality bases for downstream genotyping
The helper script fastq2a.py can be used to convert FASTQ assembly files into FASTA format while masking low-quality bases. Replacing bases below a user-defined Q-score threshold with "N" helps maintain robustness in downstream bacterial cgMLST analyses. Tools such as Ridom SeqSphere exclude alleles containing these ambiguous bases to prevent false distance calculations. However, the final count of remaining target loci should be monitored to ensure sufficient resolution for high-resolution typing. For assemblies with ~5 to 10 LQBs/Mpb, masking bases with qscores ≤10 provides a good balance between accuracy and genomic resolution.
Note: if you installed via pip/uv, you can run this helper script using:
python -m alpaqa.fastq2a -i assembly.fastq -o assembly.masked.fasta -q 10
License
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file alpaqa_bio-0.1.4.tar.gz.
File metadata
- Download URL: alpaqa_bio-0.1.4.tar.gz
- Upload date:
- Size: 21.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.2 {"installer":{"name":"uv","version":"0.11.2","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d2bccb4db18e66c7ce61b6e65694aab506a1de656394519c5230e2fd4b1fccc6
|
|
| MD5 |
00898855eb46f99c1670aea53ad08d0c
|
|
| BLAKE2b-256 |
85d638ffd433fa4237a78fbf7d31ac21ab2a0442f9a4abc042435a77e875ecb0
|
File details
Details for the file alpaqa_bio-0.1.4-py3-none-any.whl.
File metadata
- Download URL: alpaqa_bio-0.1.4-py3-none-any.whl
- Upload date:
- Size: 21.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.2 {"installer":{"name":"uv","version":"0.11.2","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
321344dded2abfcbe4aa330a1a8fb252d78c576c640a375fb9f0c1455a6d8355
|
|
| MD5 |
a0422d622fd3e4d2435de6564a559b44
|
|
| BLAKE2b-256 |
10a7ee4d28969f623c953c3812e6d3886c5563ba3aecb5869e39207a9fa9c83d
|







