A tool to use the CAMPER dataset


Project description
BETA RELEASE
The CAMPER software and data is currently in a testing state and needs more validations before it should be used for any publication or production task. This RELEASE is for early adopters to get a taste of what CAMPER can do and how it can be incorporated into a workflow. An official release will be available soon.
License Pending
This software remains unlicensed and the Wrighton Lab reserves all rights for the time being. Contributions will be welcomed once an appropriate license or licenses can be written. This should serve as another reminder that this project is in beta.
CAMPER (BETA)

Table of Contents
Overview
Curated Annotations for Microbial (Poly)phenol Enzymes and Reactions (CAMPER) is a tool that annotates genes likely involved in transforming polyphenols, and provides chemical context for these transformations in a summarized form. CAMPER aims to address a blind spot in microbial metabolism. It is currently challenging to infer polyphenol metabolism from genomic data because:
- Genes encoding characterized enzymes have not been propagated into annotation databases
- Genes in databases can often be involved in multiple pathways, requiring expert knowledge to get polyphenol context
- Polyphenols can be transformed in many ways – oxidized, reduced, demethylated, deglycosylated, etc.
- Polyphenols are a complex class of compounds
These challenges limit widespread understanding of the transformation of these compounds across environments.
To facilitate the inference of polyphenol metabolism from genomes, CAMPER includes 8 Hidden-Markov Model (HMM) profiles and 33 Basic Local Alignment Search Tool (BLAST) searches for (poly)phenol-active genes. We also provide recommended score cut-offs for searches using two ranks: a more stringent, trusted rank (A) and a more relaxed, exploratory rank (B). The development of these profiles will be described in McGivern et al (in prep). Beyond these 41 profiles, nearly 300 other annotations from other databases (KEGG, dbCAN) are included in the CAMPER summarization.
CAMPER summarizes the gene annotations into 101 modules representing different polyphenol transformations. These modules are classified by the family and sub-family of polyphenols used as substrates (following Phenol-Explorer Ontology) and by the oxygen requirements for the genes involved. These modules can be as small as a single gene, up to a maximum of 12 genes in the largest module.
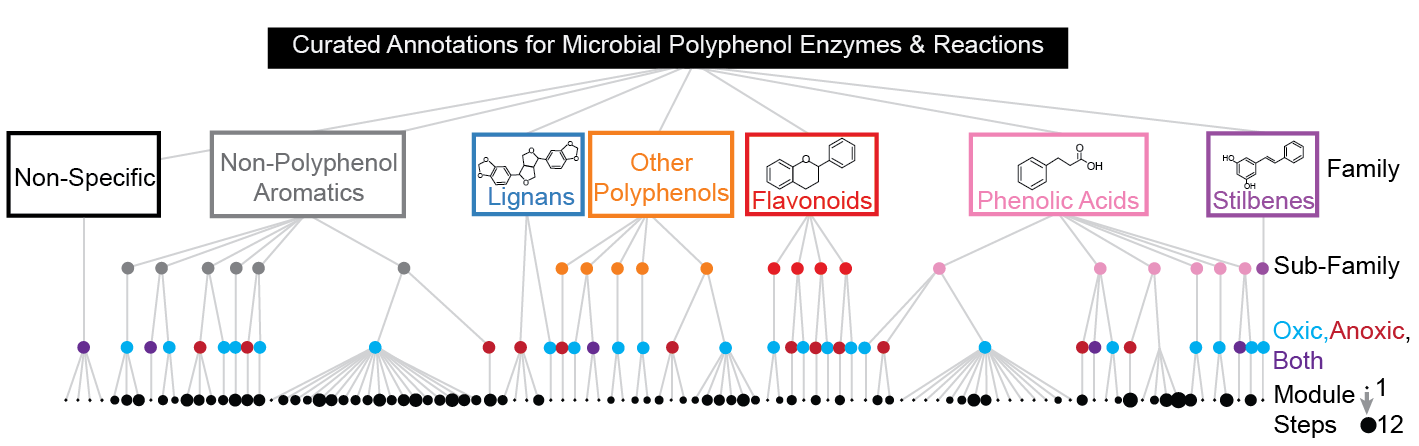
For more detailed information on the organization and outputs, see the CAMPER Outputs section below, and for module visuals, see the CAMPER Map.
Installation and Usage
There are currently three ways to run CAMPER, depending on your goals.
CAMPER DATA
The CAMPER data set consists of 5 files, each serving a key role in enabling reproducible annotation of gene data.
- CAMPER_blast.faa: A fasta file of CAMPER genes used as a target in a BLAST style search provided by mmseqs search.
- CAMPER.hmm: A HMM file used as the target in an HMM profile search provide by MMseqs profilesearch
- CAMPER_blast_scores.tsv: Provides the minimum cut off scores for search results and quality ranks with BLAST style searches.
- CAMPER_hmm_scores.tsv: Provides the minimum cut off scores for search results and quality ranks with HMM Profile searches.
- CAMPER_distillate.tsv: A custom distillate, for use with DRAM or with CAMPER_DRAMKit, to summarize the annotation results.
1. Using CAMPER within DRAM
If your goal is to integrate CAMPER into your regular genome annotation pipeline, we recommend running it as part of DRAM. You will also get the benefit of summarizing 300 annotations derived from KEGG and dbCAN databases, if you have these databases installed, in addition to the 41 CAMPER annotations. This will provide curated annotation and summarization of polyphenol transformation genes in addition to the regular DRAM databases.
There are two steps to running CAMPER in DRAM: (1) annotation and (2) summarization (distillation, in DRAM lingo).
Supply the --use_camper flag during the annotation step, like so:
DRAM.py annotate --use_camper -i 'my_bins/*.fa' -o DRAM_wCAMPER
DRAM.py distill -a DRAM_wCAMPER/annotations.tsv -o DRAM_wCAMPER_distilled
The difference in outputs between this and default DRAM is that you will find CAMPER-specific columns added to the annotations.tsv and you will find a CAMPER tab in your metabolism_summary.xlsx output.
For descriptions of the content in output files, see the CAMPER Outputs section below.
2. CAMPER Standalone Tool: CAMPER_DRAMKit
If your goal is to only look at CAMPER annotations, we recommend running CAMPER_DRAMKit, a standalone tool that provides curated annotation and summarization of polyphenol transformation genes. Unless you run with DRAM as described below and have KEGG and dbCAN databases, here you will only get the 41 custom annotations within CAMPER. CAMPER_DRAMKit is really a smaller version of DRAM that follows much the same workflow as DRAM and has similar capabilities. There are three ways to set up the CAMPER_DRAMKit:
A. Setup with Conda
The simplest way to get started with the CAMPER_DRAMKit is with Conda, using the enviroment.yaml provided in this repository.
CAMPER_DRAMKit comes with the latest version of CAMPER preloaded, so if all you want to do is annotate and distill called genes with CAMPER, you only need the following commands.
wget https://raw.githubusercontent.com/WrightonLabCSU/CAMPER/main/CAMPER_DRAMKit/environment.yaml
conda env create --name CAMPER -f ./environment.yaml
However, if you want to have all of CAMPER at your fingertips, you can use the following command to both download this repository, including all the CAMPER data and install the CAMPER_DRAMKit tool.
git clone https://github.com/WrightonLabCSU/CAMPER.git
cd CAMPER/CAMPER_DRAMKit
conda env create --name CAMPER -f ./environment.yaml
In both cases, you can activate the newly made environments with the command:
conda activate CAMPER
Provided all things have gone smoothly, you will be able to activate this environment at any time and use any of the commands outlined in the Usage section below. If there are any problems, please open an issue in the GitHub repository.
B. Setup with pip
If you are not able to use Conda, you can still install CAMPER_DRAMKit with pip using the command below. Note that first you will need to install manually install scikit-bio, and MMseqs2, as these tools can't be installed with the other pip dependencies.
pip install camper_dramkit
C. Installing with DRAM
If you intend to use CAMPER_DRAMKit with DRAM, it may be expedient to install them in the same Conda environment. This is easy to do if you have already made a DRAM Conda environment with the instructions in the DRAM README, then you can add CAMPER with the following commands:
wget https://raw.githubusercontent.com/WrightonLabCSU/CAMPER/main/CAMPER_DRAMKit/environment.yaml
conda env update --name DRAM -f ./environment.yaml
Note: If you install CAMPER_DRAMKit, you will get the latest version of the CAMPER database with it. If you want more control over the database, you can override the default data with the instructions in Other Tools and flags.
Using CAMPER_DRAMKit
Once installed, CAMPER_DRAMKit will provide three commands: camper_annotate, camper_distill and combine_annotations_lowmem. These commands alongside DRAM enable a variety of workflows.
Standalone Workflow
The simplest workflow is the two-step annotation and summarization of a single amino acid fasta file. An example of such a workflow is shown below. This will run CAMPER without any DRAM input at all, and the results will be arguably incomplete without the dram data. In order to use CAMPER_kit with DRAM see the next section.
camper_annotate -i my_genes.faa -o my_output #annotate, note the lack of the "-a" argument, this will not have dram data
camper_distill -a my_output/annotations.tsv -o my_output/distillate.tsv #summarize
These commands will make two files in the output directory (above named my_output, but this is customizable): annotations.tsv and distillate.tsv (or whatever you name it in your -o command). For descriptions of these files, see the CAMPER Outputs section below.
DRAM Combination Workflow
It is possible to use CAMPER with any version of DRAM after 1.3, with one additional command. First follow the instructions above to update your DRAM environment with CAMPER_DRAMKit. Then with that environment activated, you should be able to run the following commands to make a new raw annotations file with all the DRAM data you expect, and the CAMPER data added in.
If you are not able to update your DRAM environment for whatever reason, you will simply need to switch environments mid-workflow.
DRAM.py annotate -i 'my_bins/*.fa' -o dram_output
camper_annotate -i my_genes.faa -a dram_output_annotation/annotations.tsv -o camper_dram_output
This will create a new set of raw annotations with CAMPER data added, in this case the path of the new file will be camper_dram_output/annotations.tsv. Then, use the camper_distill command to get a distillate with all the key genes from both DRAM and CAMPER.
camper_distill -a camper_dram_output/annotations.tsv -o camper_dram_output/camper_distillate.tsv
For descriptions of the annotations.tsv and summary file, see the CAMPER Outputs section below.
Other Tools and flags
In order to further customize your workflow, you can take advantage of a few more options in the CAMPER_DRAMKit package:
Combine Annotations With low memory: You may want to re-annotate many existing DRAM annotation files, possibly from more than one version of DRAM. To this end we include the combine_annotations_lowmem command which should combine many annotation files quickly and with a small memory footprint, even if they come from different versions of DRAM. The command is used like so:
Combine_annotations_lowmem -i /path/to/many/dramfolders/*/annotations.tsv -o combined_annotation.tsv
The input path needs to be a wild card pointing to a set of DRAM annotation files, this is passed to the python glob command, but the format should be familiar to anyone who uses bash and you can test it with the ls command.
Manually Specifying the Location of CAMPER Files: The behavior of the camper_annotate and camper_distill commands is controlled by the latest version of the CAMPER dataset. If you want to use an older version of CAMPER, it is suggested you install the older version of the CAMPER_DRAMKit tool, as they will be released together and be mutually compatible. However, if you must, you can also specify the files to use with camper_annotate and camper_distill using the appropriate arguments. An example is shown below.
camper_annotate -i my_genes.faa -o my_output \
--camper_fa_db_loc CAMPER_blast.faa \
--camper_fa_db_cutoffs_loc CAMPER_blast_scores.tsv \
--camper_hmm_loc CAMPER.hmm \
--camper_hmm_cutoffs_loc CAMPER_hmm_scores.tsv
camper_distill -a my_output/annotations.tsv -o my_output/distillate.tsv \
--camper_distillate CAMPER_distillate.tsv
If at any time you forget these arguments, remember that running any script with the --help flag will provide more information. Also note that if you do not specify one or more arguments, the default data will be used.
3. I just want to run your BLAST and HMM searches on my own!
We get that sometimes this is all you want to do! This is the simplest way to use our annotations. See the above CAMPER Data section and download the CAMPER_blast.faa and CAMPER.hmm files. These can be run using blast, hmmsearch, or mmseqs2 searches of your data, for example:
makeblastdb -in CAMPER_blast.faa -dbtype prot
blastp -query my_genes.faa -db CAMPER_blast.faa -out BLAST_my_genes_CAMPER.txt -outfmt 6
hmmsearch --tblout hmmsearch_my_genes_CAMPER.txt CAMPER.hmm my_genes.faa
We strongly recommend curating these outputs with the scores given in the CAMPER_blast_scores.tsv and CAMPER_hmm_scores.tsv files for each profile search.
CAMPER Outputs
Approaches 1 and 2 output two files: the raw information for given searches (annotations.tsv) and the summarized information across searches (the distillate, either the metabolism_summary.xlsx if run through DRAM or the distillate.tsv from CAMPER_DRAMKit).
Raw annotations: This is either a standalone file, or columns added to a file, depending on the search approach. This file tells you the genes in your dataset that pass CAMPER annotation thresholds, what they are annotated as, and the scores. It includes the following columns:
camper_hits, A longer ID giving the CAMPER ID, gene abbreviation, and gene description.camper_rank, A match quality rank based on the value of the bit score (A or B). For BLAST-style searches, an A rank is a bitscore >=200 and B >=120. For HMM-style searches, scores are specific to each profile (seeCAMPER_hmm_scores.tsv).camper_bitScore, The bitscore from the best search result. If more than one search meets at least a B-rank for a given gene, the search with the higher score is reported.camper_id: Unique CAMPER ID used in the distillation step, of the form D000XX.camper_definition: A short description of the CAMPER match in the database.camper_search_type: Tells you if a HMM profile or blast search found this match.
Distillate: This is either a single file, or the CAMPER tab in the metabolism_summary.xlsx file. Each row in this file corresponds to a gene in a CAMPER module. This file gives you gene counts of genes in CAMPER modules. It includes the following columns:
gene_id, the database IDs assigned to this gene. These can be from CAMPER (D000XX), KEGG (KXXXX), dbCAN (AAX), or EC numbers. Note, some IDs are included more than once in the sheet if they are involved in more than one module!gene_description, A more informative description of the gene in the step, including gene abbreviation and gene name.module, The CAMPER module that the given gene belongs to. There are 101 modules in CAMPER.header, The classification for the polyphenol substrate following Phenol-Explorer Ontology. In the form: Polyphenol;Family;Sub-Family;Compound.subheader, This contains information about routes, steps, and subunits. Sometimes, a given transformation can be accomplished in more than one sequence of steps: these are termed 'Routes'. Steps indicate the sequential transformations in the module. Subunits denote if the given gene encodes a subunit of a larger complex that carries out a step. Sometimes steps are labeled as "optional" if they are not required.specifc_reaction, This gives examples of reactions when possible.oxygen, This is either "oxic", "anoxic"," or "both" for reactions that require oxygen, don't require oxygen, or can function with or without, respectively. Note: these are largely based on literature reporting and the systems they were characterized in, and should be used as guidelines.EC, The EC number (if known) for a reaction.Notes, Any important information to know about the genes, for example: manual curation to do, note on gene clusters, should they be extracellular etc. The remaining columns will be counts of each gene in your input files.
CAMPER Map
This is also provided as a PDF file.
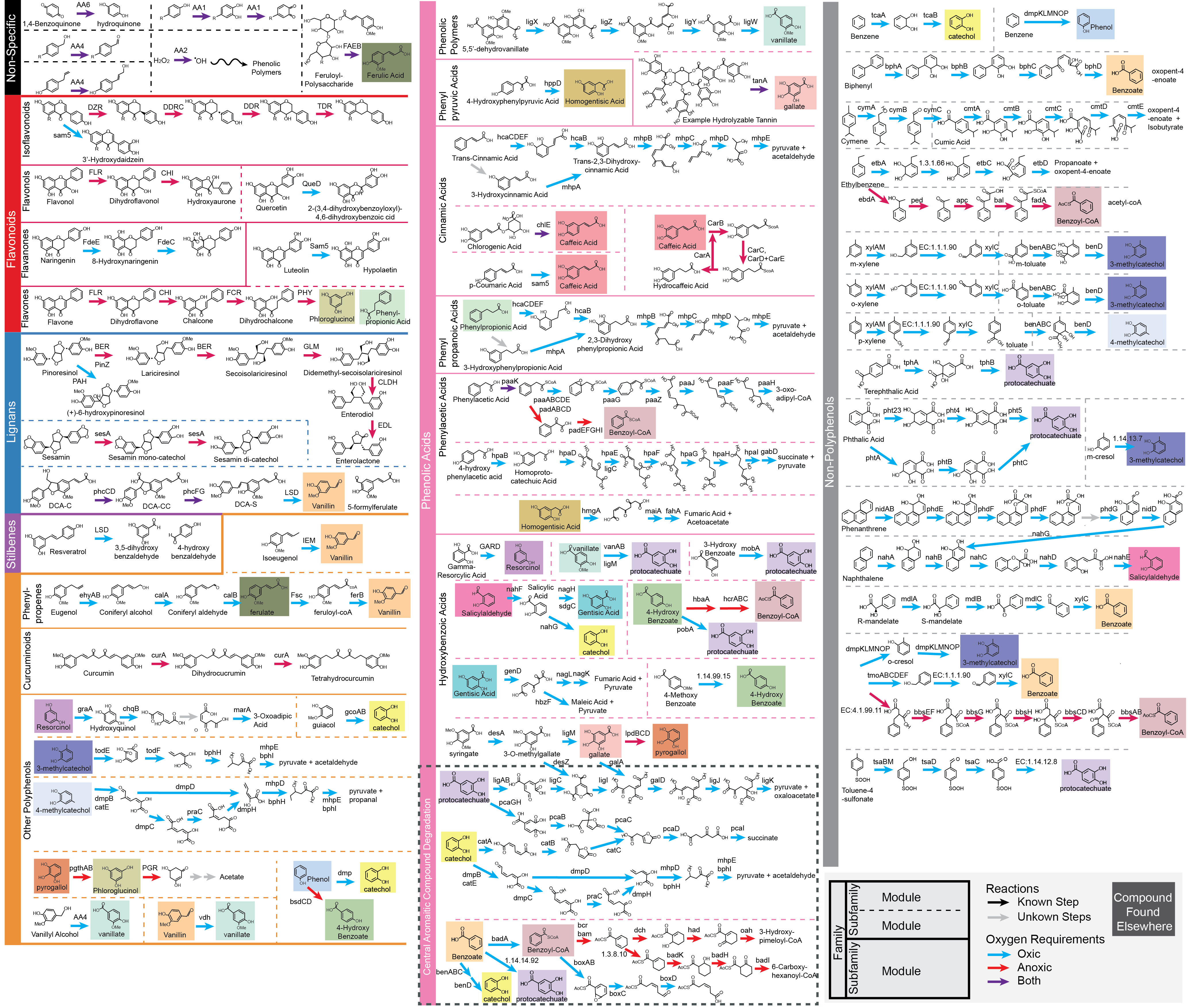
Happy CAMPER-ing!
Annotations, organization, and conceptualization by Bridget McGivern. Coding and implementation by Rory Flynn.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file camper_dramkit-1.0.12.tar.gz.
File metadata
- Download URL: camper_dramkit-1.0.12.tar.gz
- Upload date:
- Size: 1.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.10.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
98c2a020f51ba0bec1cf1bb5e2020e5ce6898889ce2284960435cea70164ad3f
|
|
| MD5 |
a87eb7bbc75f321a00ea7b73eaff0343
|
|
| BLAKE2b-256 |
42b6b9b9c6e0c85843a01eb211f28161d75524deb6beeb9f8ecc5098678acdcf
|







