A library to combine, analyze, group and correct p-values in BED files. Unique tools involve correction for spatial autocorrelation. This is useful for ChIP-Seq probes and Tiling arrays, or any data with spatial correlation.
About
- The Bioinformatics Applications Note manuscript is available here:
http://bioinformatics.oxfordjournals.org/content/28/22/2986.full
It includes an explanation of 3 examples in the examples directory of this repository.
The software is distributed under the MIT license.
QuickStart

If your data is a sorted BED (first columns are chrom, start, stop) with a column for p-value in the 4th column from single-probe tests–e.g. from limma::topTable(…, n=Inf), you can find DMRs as:
comb-p pipeline \
-c 4 \ # p-values in 4th column
--seed 1e-3 \ # require a p-value of 1e-3 to start a region
--dist 200 # extend region if find another p-value within this dist
-p $OUT_PREFIX \
--region-filter-p 0.1 \ # post-filter reported regions
--anno mm9 \ # annotate with genome mm9 from UCSC
$PVALS # sorted BED file with pvals in 4th column
The output will look like:
https://github.com/brentp/combined-pvalues/blob/master/manuscript/anno.tsv
With DMRs annotated to the nearest gene and CpG island. Negative distances indicate that the DMR is upstream of the gene. DMRs inside of genes have exon / UTR or the appropriate feature to indicate their location within the gene. If matplotlib is installed, then you will get a figure like this:
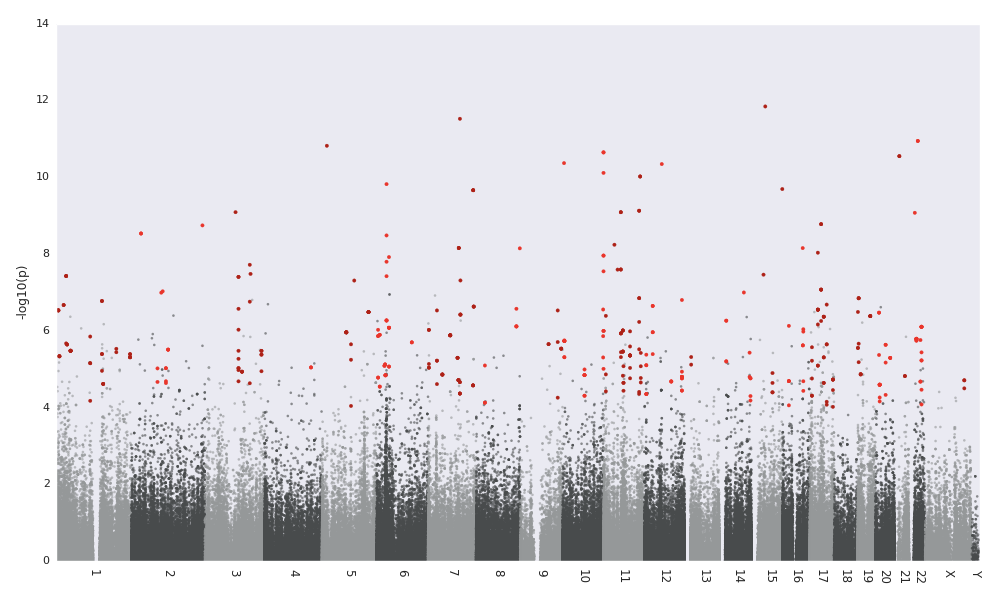
Manhattan plot of p-values with DMRs highlighted
Regions passing the –region-filter-p are highlighted in a red color.
Commands below give finer control over each step.
Installation
comb-p requires python2.7 or python3.7+
If you do not have numpy and scipy installed. Please use anaconda from: http://continuum.io/downloads which is a complete python distribution with those modules included.
run:
sudo python setup.py install
to have comb-p installed on your path. Otherwise, you can use the python scripts in the cpv subdirectory. E.g.
python cpv/peaks.py
corresponds to the command:
comb-p peaks
Invocation
The program is run with:
$ comb-p
This message is displayed:
To run, indicate one of: acf - calculate autocorrelation within BED file slk - Stouffer-Liptak-Kechris correction of spatially correlated p-values fdr - Benjamini-Hochberg correction of p-values peaks - find peaks in a BED file. region_p - generate SLK p-values for a region (of p-values) manhattan - a manhattan plot of values in a BED file. pipeline - run the series of commands to find DMRs. NOTE: most of these assume *sorted* BED files.
Where each of the listed modules indicates an available program. Running any of the above will result in a more detailed help message. e.g.:
$ comb-p acf -h
Gives:
usage: comb-p [-h] [-d D] [-c C] files [files ...]
calculate the autocorrelation of a *sorted* bed file with a set
of *distance* lags.
positional arguments:
files files to process
optional arguments:
-h, --help show this help message and exit
-d D start:stop:stepsize of distance. e.g. 15:500:50 means check acf
at distances of:[15, 65, 115, 165, 215, 265, 315, 365, 415, 465]
-c C column number with p-values for acf calculations
Indicating that it can be run as:
$ .comb-p acf -d 1:500:50 -c 5 data/pvals.bed > data/acf.txt
Each module is described in detail below.
Example
Find and merge peaks/troughs within a bed file
python cpv/peaks.py --seed 0.05 --dist 1000 data/pvals.bed > data/pvals.peaks.bed
This will seed peaks with values < 0.05 and merge any adjacent values within 1KB. The output is a BED file containing the extent of the troughs. If the argument –invert is specified, the program will find look for values larger than the seed.
Pipeline
The default steps are to:
calculate the ACF
use the ACF to do the Stouffer-Liptak correction
do the Benjamini-Hochberg FDR correction
find regions from the adjusted p-values.
Inputs and outputs to each step are BED files.
Note that any of these steps can be run independently, e.g. to do multiple testing correction on a BED file with p-values, just call the fdr.py script.
ACF
To calclulate autocorrelation from 1 to 500 bases with a stepsize of 50 on the p-values in column 5, the command would look something like:
$ python cpv/acf.py -d 1:500:50 -c 5 data/pvals.bed > data/acf.txt
The ACF will look something like:
# {link}
lag_min lag_max correlation N
1 51 0.06853 2982
51 101 0.04583 4182
101 151 0.02719 2623
151 201 0.0365 3564
201 251 0.0005302 2676
251 301 0.02595 3066
301 351 0.04935 2773
351 401 0.04592 2505
401 451 0.03923 2972
Where the first and second columns indicate the lag-bin, the third is the autocorrelation at that lag, and the last is the number of pairs used in calculating the autocorrelation. If that number is too small, the correlation values may be unreliable. We expect the correlation to decrease with increase lag (unless there is some periodicity).
That output should be directed to a file for use in later steps.
Combine P-values with Stouffer-Liptak-Kechris correction
See
The ACF output is then used to do the Stouffer-Liptak-Kechris correction. A call like:
$ python cpv/slk.py --acf data/acf.txt -c 5 data/pvals.bed > data/pvals.acf.bed + adjusts the p-values by stouffer-liptak with values from the autocorrelation in the step above. + outputs a new BED file with columns:
chr, start, end, pval, combined-pval
Regions
We are often interested in entire regions. After running the above example, we can find the extent of any regions using:
$ python cpv/peaks.py --dist 500 --seed 0.1 \
data/pvals.adjusted.bed > data/pvals.regions.bed
where the seed inidicates a minimum p-value to start a region. Again, -c can be used to indicate the column containing the p-values (defaults to last column)`–dist` tells the program to merge peaks (in this case troughs) within 500 bases of the other. The output file is a BED file with each region and the lowest (currently) p-value in the region.
The cpv/peaks.py script is quite flexible. Run it without arguments for further usage.
Region P-values (region_p)
The reported p-value is a Stouffer-Liptak p-value for the entire region. This is done by taking a file of regions, and the original, uncorrected p-values, calculating the ACF out to the length of the longest region, and then using that ACF to perform the Stouffer-Liptak correction on each region based on the original p-values. The 1-step Sidak correction for multiple testing is performed on the p-value for the region. Because the original p-values are sent in, we know the coverage of the input. The Sidak correction is then based on the number of possible regions of the current size that could be created from the total coverage. The extra columns added to the output file are the Stouffer-Liptak p-value of the region and the Sidak correction of that p-value.
An invocation:
$ comb-p region_p -p data/pvals.bed \
-r data/regions.bed \
-s 50 \
-c 5 > data/regions.sig.bed
Will extract p-values from column 5 of pvals.bed for lines within regions in regions.bed.
Frequently Asked Questions
See the Wiki F.A.Q.
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file cpv-0.50.4.tar.gz.
File metadata
- Download URL: cpv-0.50.4.tar.gz
- Upload date:
- Size: 68.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.5.0.1 requests/2.23.0 setuptools/47.1.1.post20200529 requests-toolbelt/0.9.1 tqdm/4.46.1 CPython/3.7.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
65feec1f426372f4be77c3d14046a4521aff4f2c827f47af2e2491c4ee1db156
|
|
| MD5 |
47d73d6a30489f92b31857215828cb7f
|
|
| BLAKE2b-256 |
ea4c86b410772d5aadb2b7247648cde16a35412b78b7a7a815cfd76465abf7aa
|









