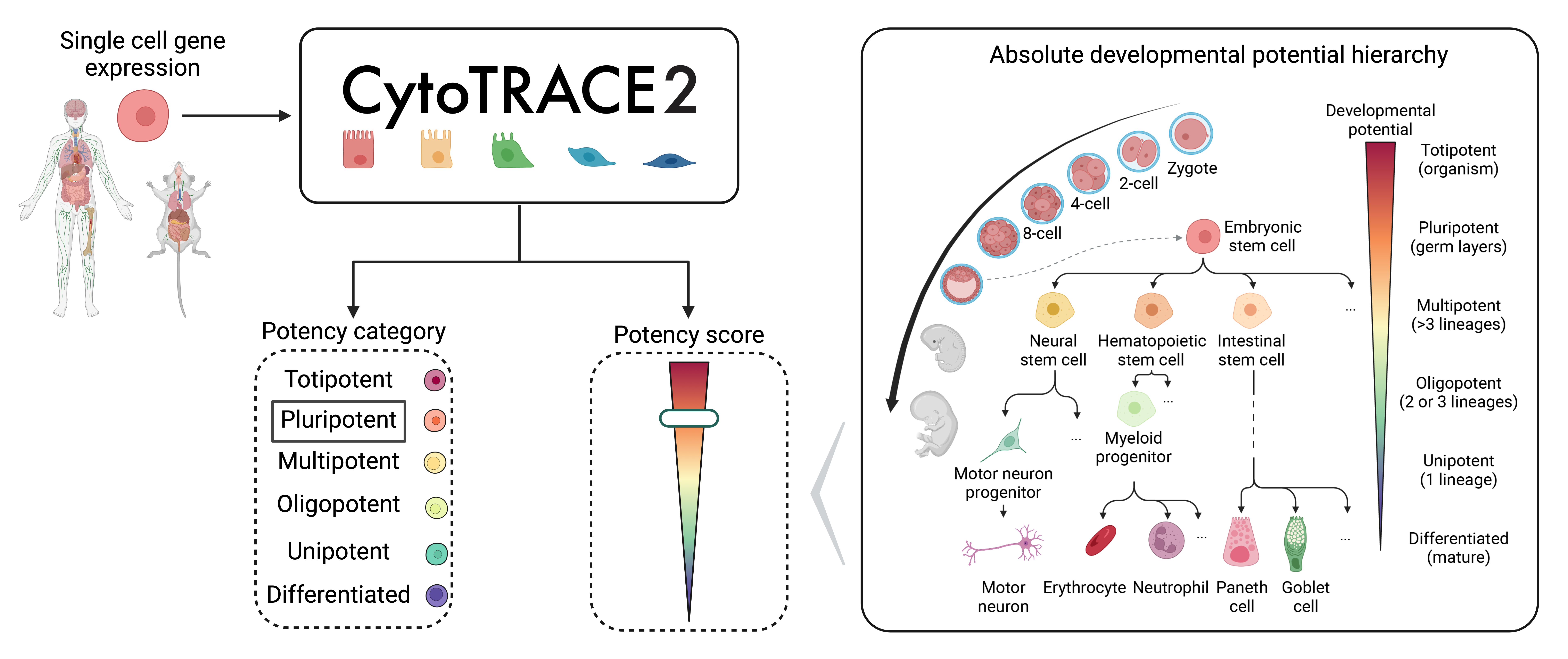
CytoTRACE 2 is a computational method for predicting cellular potency categories and absolute developmental potential from single-cell RNA-sequencing data.

Project description

Prediction of absolute developmental potential
using single-cell expression data
CytoTRACE 2 is a computational method for predicting cellular potency categories and absolute developmental potential from single-cell RNA-sequencing data.
Potency categories in the context of CytoTRACE 2 classify cells based on their developmental potential, ranging from totipotent and pluripotent cells with broad differentiation potential to lineage-restricted oligopotent, multipotent and unipotent cells capable of producing varying numbers of downstream cell types, and finally, differentiated cells, ranging from mature to terminally differentiated phenotypes.
The predicted potency scores additionally provide a continuous measure of developmental potential, ranging from 0 (differentiated) to 1 (totipotent).
Underlying this method is a novel, interpretable deep learning framework trained and validated across 34 human and mouse scRNA-seq datasets encompassing 24 tissue types, collectively spanning the developmental spectrum.
This framework learns multivariate gene expression programs for each potency category and calibrates outputs across the full range of cellular ontogeny, facilitating direct cross-dataset comparison of developmental potential in an absolute space.
Installation instructions
Expand section
Install using pip/pip3:
pip install cytotrace2-py
Optional:
For faster data reading, you can optionally install datatable (e.g., pip install datatable or conda install -c conda-forge datatable) if your OS and environment support it. If not installed, the code will default to using pandas for reading input files.
Input files
Expand section
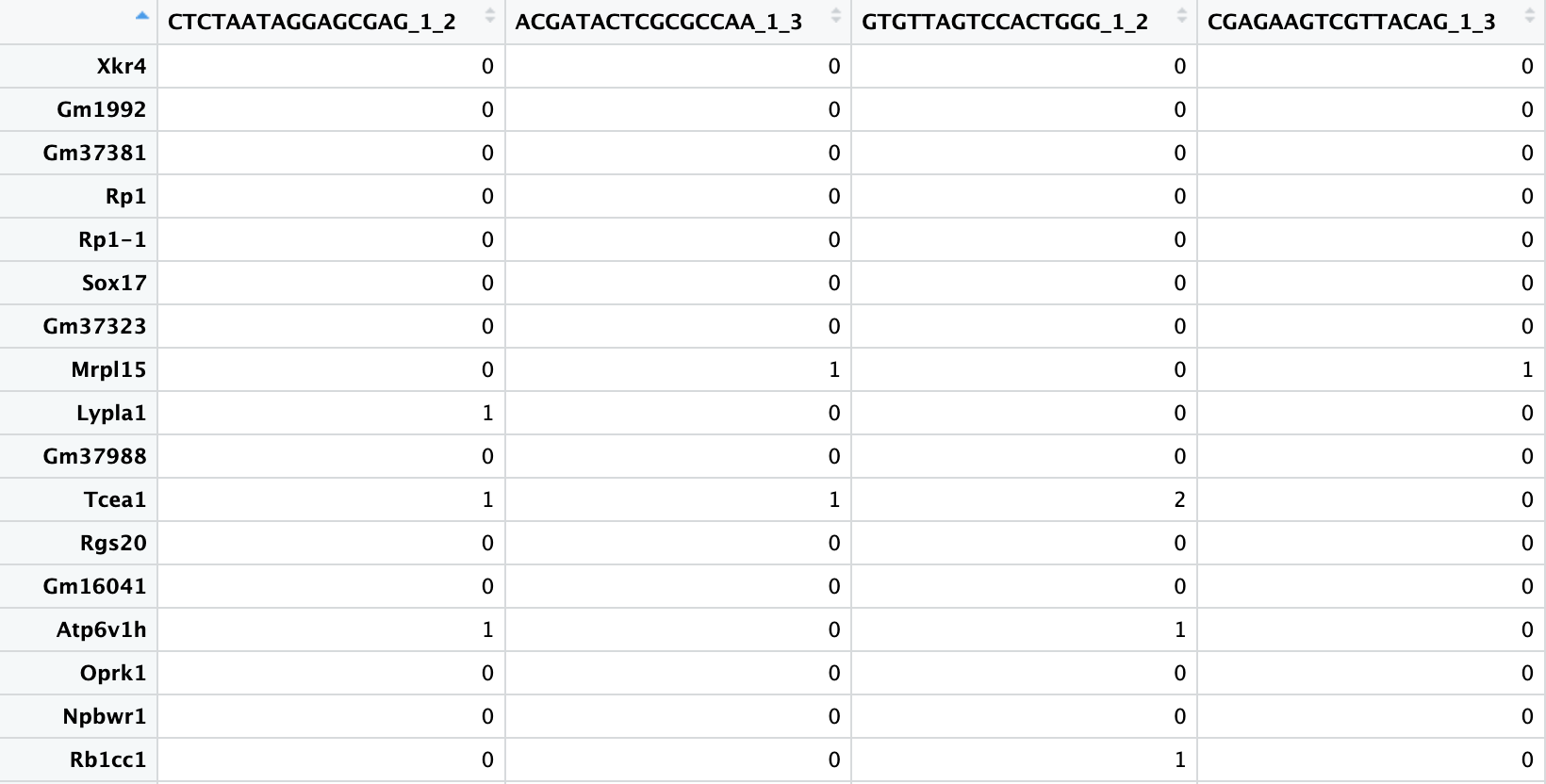
By default, CytoTRACE 2 requires only a single-cell gene expression file as input. For output plotting, a cell annotation file is accepted as well. All files should be provided in a tab-delimited tabular input format (.txt) with no double quotations. Further formatting details for each input file are specified below.
scRNA-seq gene expression file:
- The table must be genes (rows) by cells (columns).
- The first row must contain the single cell IDs and the first column must contain the gene names.
- The gene expression data can be represented as raw or normalized counts, as long as normalization preserves relative ranking of input gene values within a cell.
- No instances of duplicate gene or cell names should be present.
Cell annotation file:
- The table should contain two columns, where column 1 contains the single cell IDs corresponding to the columns of the scRNA-seq matrix and column 2 contains the corresponding cell type annotations.
- The columns must have a header.
- Additional columns beyond the first two will be ignored.

Running CytoTRACE 2
Expand section
After installing the package and its dependencies, the tool is ready to run both from command line and from within a Python script.
NOTE: Only during the first run, the script will download remotely hosted underlying models. This is done just once and saved for all future runs.
For mouse data with default settings, CytoTRACE 2 would be run from command line as:
cytotrace2 \
--input-path /path/to/cell_expression \
--annotation-path /path/to/cell_annotations \
--species mouse
Or with more condensed parameter names:
cytotrace2 \
-f /path/to/cell_expression \
-a /path/to/cell_annotations \
-sp mouse
CytoTRACE 2 can also be called from within a Python script after importing the package.
from cytotrace2_py.cytotrace2_py import *
input_path = "/path/to/cell_expression.txt"
example_annotation_path = "/path/to/cell_annotations.txt"
example_species = "human"
results = cytotrace2(input_path,
annotation_path=example_annotation_path,
species=example_species)
NOTE: When running on computers with less than 16GB memory, we recommend reducing max_cores parameter to 1 or 2 to avoid memory issues.
Extended usage details
Expand section
Key notes:
- By default, CytoTRACE 2 expects mouse data. To provide human data, users should specify
species = "human"
Required input:
- input_path: A filepath to a .txt file containing a single-cell RNA-seq gene expression matrix (rows as genes, columns as cells)
Optional arguments:
- annotation_path: A filepath to a .txt file containing phenotype annotations corresponding to the cells of the scRNA-seq expression matrix
- species: String indicating the species name for the gene names in the input data (options: "human" or "mouse", default is "mouse").
- batch_size: Integer indicating the number of cells to process at once for the pipeline steps, including subsampling for KNN smoothing (default is 10000).
- smooth_batch_size: Integer indicating the number of cells to subsample further within the batch_size for the smoothing by diffusion step of the pipeline (default is 1000).
- disable_parallelization: Flag indicating whether to disable parallelization (default is FALSE, or absent for the command line).
- disable_plotting: Flag indicating whether to disable plotting (default is FALSE, or absent for the command line). To plot results, data are reprocessed following the core CytoTRACE 2 pipeline to produce UMAP embeddings via a standard scanpy pipeline. As this step can be time-consuming, we provide the option to disable it if desired.
- max_cores: Integer indicating user-provided restriction on the maximum number of CPU cores to use for parallelization (default is None, and the number of cores used will then be determined based on system capacity; when running on computers with less than 16GB memory, we recommend reducing it to 1 or 2 to avoid memory issues.).
- seed: Integer specifying the seed for reproducibility in random processes (default is 14).
- output_dir: Path to the directory to which to save CytoTRACE 2 outputs (default is cytotrace2_results in the current working directory).
Information about these arguments is also available in the function's manual, which can be accessed by running cytotrace2 -h from the command line.
For more information about the tool, R/Python package documentation, Vignettes, example input files, and more, visit out GitHub page: https://github.com/digitalcytometry/cytotrace2.
CytoTRACE 2 web application
An interactive RShiny web application can be accessed at cytotrace2.stanford.edu, allowing users to run analyses on their own data, browse results for 33 ground-truth–annotated datasets, explore potency-associated genes and gene-set enrichment across the single-cell potency atlas, download the atlas, and access Python vignettes for model training and custom GSBN architectures.
Authors
CytoTRACE 2 was developed in the Newman Lab by Minji Kang, Gunsagar Gulati, Erin Brown, Susanna Avagyan, Jose Juan Almagro Armenteros and Rachel Gleyzer.
Contact
If you have any questions, please contact the CytoTRACE 2 team at cytotrace2team@gmail.com.
License
Please see the LICENSE file.
Citation
If you use CytoTRACE 2, please cite:
Improved reconstruction of single-cell developmental potential with CytoTRACE 2. Nature Methods, 2025.
Minji Kang*, Gunsagar S. Gulati*, Erin L. Brown*, Zhen Qi*, Susanna Avagyan, Jose Juan Almagro Armenteros, Rachel Gleyzer, Wubing Zhang, Chloé B. Steen, Jeremy Philip D’Silva, Janella Schwab, Michael F. Clarke, Aadel A. Chaudhuri, and Aaron M. Newman†. doi.org/10.1038/s41592-025-02857-2


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file cytotrace2_py-1.1.0.4.tar.gz.
File metadata
- Download URL: cytotrace2_py-1.1.0.4.tar.gz
- Upload date:
- Size: 4.4 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.11.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
133f5075983ba1c4ffa0db98a46a24ad695a251bdcf5d12e5f3f3e24e2fc5589
|
|
| MD5 |
1978fa2ac3f319e5f8f9a17dd70cb5c1
|
|
| BLAKE2b-256 |
32620810b83be3f23cd14c84f9174b798ab73d2ae067d58e5dba930d851102da
|
File details
Details for the file cytotrace2_py-1.1.0.4-py3-none-any.whl.
File metadata
- Download URL: cytotrace2_py-1.1.0.4-py3-none-any.whl
- Upload date:
- Size: 4.5 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.11.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1471fbe346f10502599e894d696184067c893ff6a79ab24be6cc0c3435e4cabe
|
|
| MD5 |
5a14e07216c6947bf1f1eb24c564fa3f
|
|
| BLAKE2b-256 |
745aa439282d22b898d02a5c324ead630248a2df44309be1b6e9653d37430937
|







