Python package for fCAT, a feature-aware completeness assessment tool

Project description
fCAT




One of the critical steps in a genome sequencing project is to assess the completeness of the predicted gene set. The standard workflow starts with the identification of a set of core genes for the taxonomic group, in which the target species belongs to. The fraction of missing core genes serves then as a proxy of the target gene set completeness.
fCAT is a feature-aware Completeness Assessment Tool, that helps to answer the question "How complete is my gene set?". In particularly, fCAT checks for the presence of conserved genes (the core genes) of a specific taxonomy clade in the target gene set using feature-aware directed ortholog search fDOG. In addition to the length criteria for classifying the found orthologs (as same as BUSCO), fCAT utilizes the domain architecture similarity FAS scores to further validate the orthologs. The later gives an alternative view on the accuracy of the target gene models, which shows how different the target orthologs in comparison to the core genes in their domain architecture.
fCAT outputs both the summary result in a tabular text file and the phylogenetic profile of the core genes, which can be visualized using the tool PhyloProfile. By analyzing the profiles of the entire orthologous groups within a specific taxonomy clade, we can further identify and ultimately correct erroneous gene annotations.
Click here for the full PDF version of the ECCB2022 poster
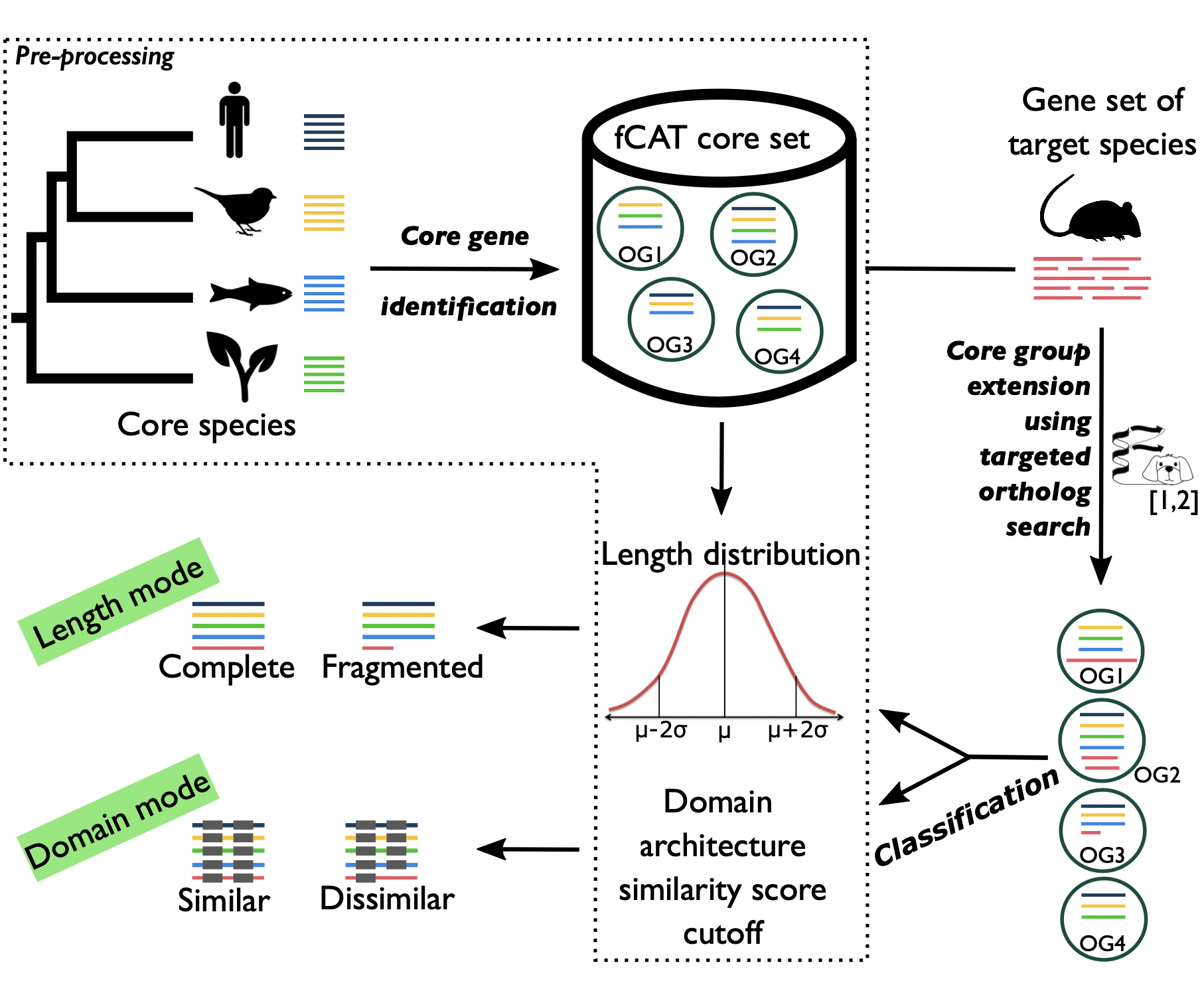
Table of Contents
How to install
fCAT tool is distributed as a python package named fcat.
Requirements
Please ensure both Python and R are properly installed and accessible in your system environment before installing fCAT.
Using a Conda Environment
-
Follow this instruction to install Mamba or Micromamba (faster package manager for conda)
-
Create a new environment
mamba create -n fcat python r-base -c conda-forge -c bioconda -c defaults -y
- Activate the environment
mamba activate fcat
- Install fcat using
pip:
python3 -m pip install fcat
Without conda
You can install fcat using pip:
python3 -m pip install fcat
or, in case you do not have admin rights, and don't use package systems like Anaconda to manage environments you need to use the --user option:
python3 -m pip install --user fcat
and then add the following line to the end of your ~/.bashrc or ~/.bash_profile file, restart the current terminal to apply the change (or type source ~/.bashrc):
export PATH=$HOME/.local/bin:$PATH
Note: fCAT requires R to be present! Please make sure that you have R installed on your computer.
Usage
fCAT depends on the following external tools:
Please make sure that both fDOG and FAS are successfully setup before using fCAT.
The complete process of fCAT can be done using one function fcat
fcat --coreDir /path/to/fcat_data --coreSet eukaryota --refspecList "HOMSA@9606@2" --querySpecies /path/to/query.fa [--annoQuery /path/to/query.json] [--outDir /path/to/fcat/output]
where eukaryota is name of the fCAT core set (equivalent to BUSCO set); HOMSA@9606@2 is the reference species from that core set that will be used for the ortholog search; query is the name of species of interest. If --annoQuery not specified, fCAT fill do the feature annotation for the query proteins using FAS tool.
Output
You will find the output in the /path/to/fcat/output/fcatOutput/eukaryota/query/ folder, where /path/to/fcat/output/ could be your current directory if you not specified --outDir when running fcat. The following important output files/folders can be found:
- all_summary.txt: summary of the completeness assessment using all 4 score modes
- all_full.txt: the complete assessment of 4 score modes in tab delimited file
- fdogOutput.tar.gz: a zipped file of the ortholog search result
- mode_1, mode_2, mode_3 and mode_4: detailed output for each score mode
- phyloprofileOutput: folder contains output phylogenetic profile data that can be used with PhyloProfile tool
Besides, if you have already run fCAT for several query taxa with the same fCAT core set, you can find the merged phylogentic profiles for all of those taxa within the corresponding core set output (e.g. /path/to/fcat/output/fcatOutput/eukaryota/*.phyloprofile).
fCAT score modes
The table below explains how the specific ortholog group cutoffs for each fCAT core set were calculated, and which value of the query ortholog is used to assess its completeness, or more precisely, its functional equivalence to the ortholog group it belongs to. If the value of a query ortholog is not less than its ortholog group cutoff, that group will be evaluated as similar or complete. In case co-orthologs have been predicted, the assessment for the core group will be duplicated. Depending on the value of each single ortholog, a duplicated group can be seen as duplicated (similar) or duplicated (dissimilar) in the full report (e.g. all_full.txt).
| Score mode | Cutoff | Value used for comparing |
|---|---|---|
| Mode 1 - Strict mode | Mean of FAS scores between all core orthologs | Mean of FAS scores between query ortholog and all core proteins |
| Mode 2 - Reference mode | Mean of FAS scores between refspec and all other core orthologs | Mean of FAS scores between query ortholog and refspec protein |
| Mode 3 - Relaxed mode | The lower bound of the confidence interval calculated by the distribution of all-vs-all FAS score in a core group | Mean of FAS scores between query ortholog and all core proteins |
| Mode 4 - Length mode | Mean and standard deviation of all core protein lengths | Length of query ortholog |
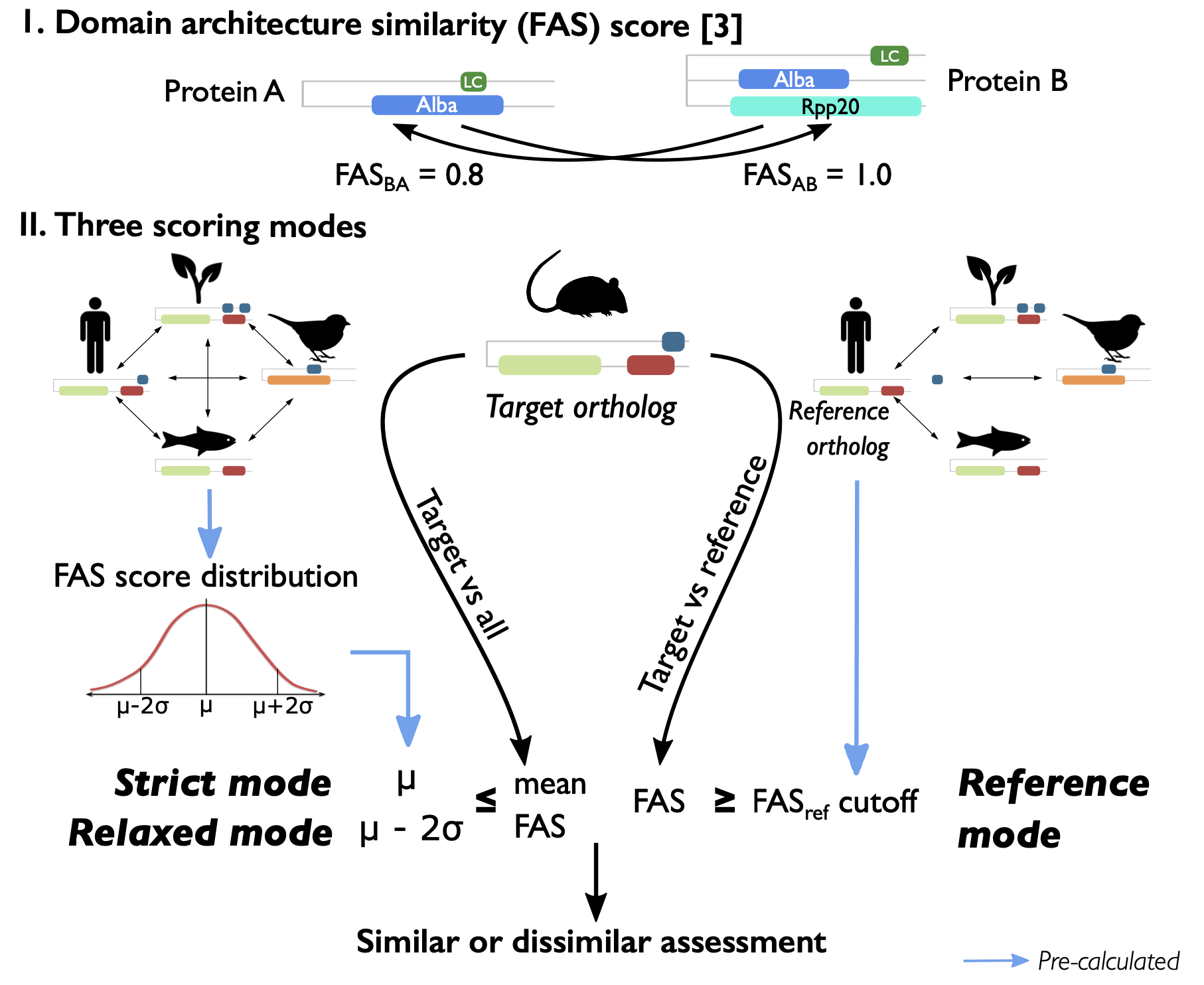
Note: FAS scores are bidirectional FAS scors; core protein or core ortholog is protein in the core ortholog groups; query protein or query ortholog is ortholog protein of query species; refspec is the specified reference species
Bugs
Any bug reports or comments, suggestions are highly appreciated. Please open an issue on GitHub or be in touch via email.
Contributors
How-To Cite
Tran V and Ebersberger I. fCAT: Assessing gene set completeness using domain-architecture aware targeted ortholog searches. F1000Research 2022, 11:1091 (poster) (doi: 10.7490/f1000research.1119126.1)
Contact
For further support or bug reports please contact: tran@bio.uni-frankfurt.de
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file fcat-0.1.11.tar.gz.
File metadata
- Download URL: fcat-0.1.11.tar.gz
- Upload date:
- Size: 39.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5f0afeaa195a77f9d09b4d731e7cd9a50dfc4972719e288792f34c57ef8be976
|
|
| MD5 |
39e6c9325915163990cc32ad331e8c3f
|
|
| BLAKE2b-256 |
5f996aa48ef0367ce6bc1871366915e0b9955a99a27b2dfaf57d9fce95e3a459
|
File details
Details for the file fcat-0.1.11-py3-none-any.whl.
File metadata
- Download URL: fcat-0.1.11-py3-none-any.whl
- Upload date:
- Size: 43.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
28b35e582e0e085b15416ea04b92b09975446957afb78d138c606a40947a2779
|
|
| MD5 |
ab07ff436271cebf7d7c50a40806fdc7
|
|
| BLAKE2b-256 |
26d2c8d30ac9a25dc3eb23c79994dcde605d09c69b3e9a88643d02c8f9244b9a
|







