A command line tool that makes it easier to find sequencing data from SRA / GEO / ENCODE / ENA / EBI-EMBL / DDBJ / Biosample.


Project description
ffq







! NCBI is deprecating .SRA file links. This may result in an empty list with `--ncbi`.
+ Have a cool use case for ffq? Submit a PR to the `Use cases` section and we'll feature it!
Fetch metadata information from the following databases:
- GEO: Gene Expression Omnibus,
- SRA: Sequence Read Archive,
- EMBL-EBI: European Molecular BIology Laboratory’s European BIoinformatics Institute,
- DDBJ: DNA Data Bank of Japan,
- NIH Biosample: Biological source materials used in experimental assays,
- ENCODE: The Encyclopedia of DNA Elements.
ffq receives an accession and returns the metadata for that accession as well as the metadata for all downstream accessions following the connections between GEO, SRA, EMBL-EBI, DDBJ, and Biosample. If you use ffq in a publication, please the cite*:
Gálvez-Merchán, Á., et al. (2022). Metadata retrieval from sequence databases with ffq. bioRxiv 2022.05.18.492548.
The manuscript is available here: https://doi.org/10.1101/2022.05.18.492548.
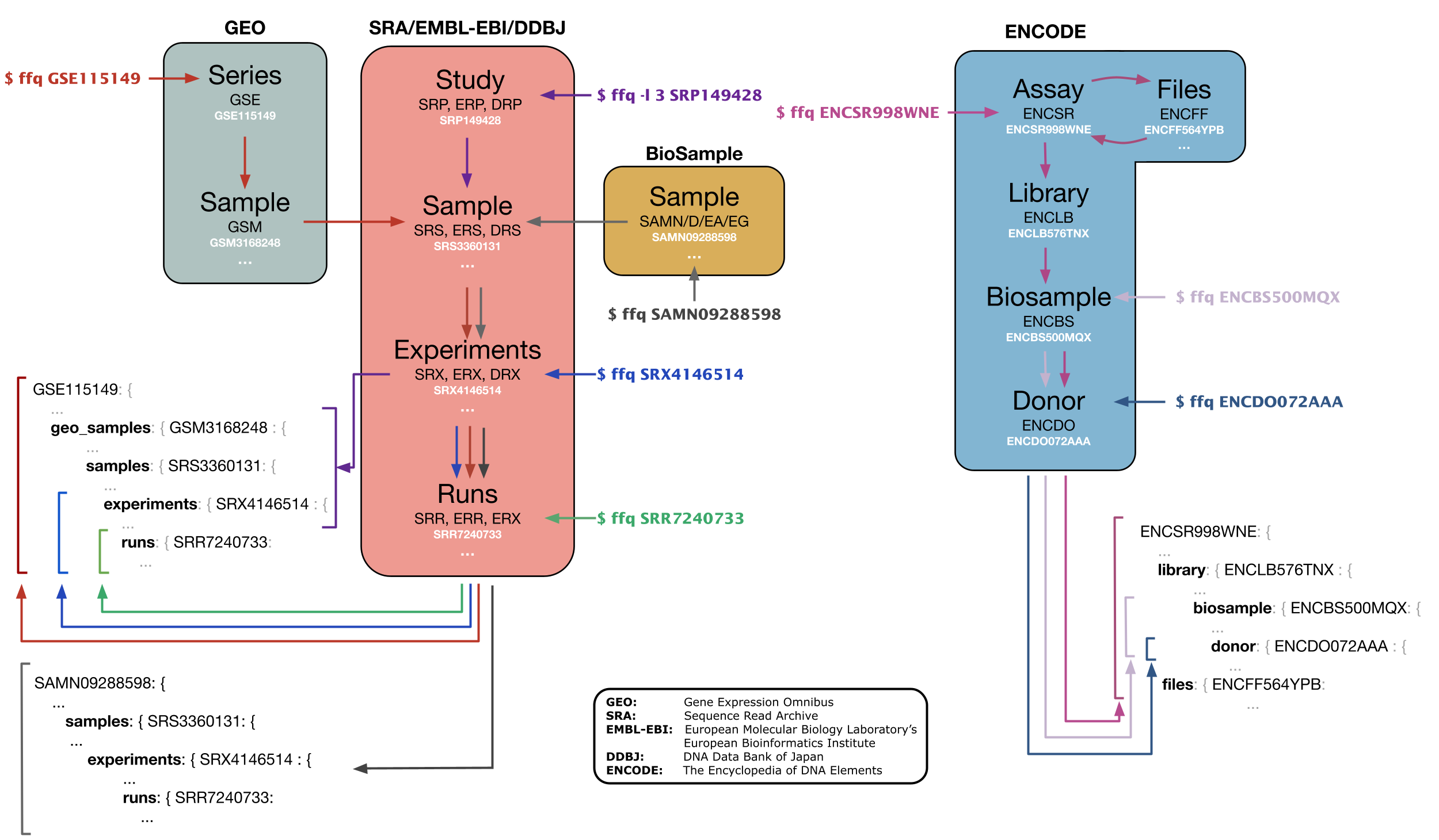
By default, ffq returns all downstream metadata down to the level of the SRR record. However, the desired level of resolution can be specified.
ffq can also skip returning the metadata, and instead return the raw data download links from any available host (FTP, AWS, GCP or NCBI) for GEO and SRA ids.
Installation
The latest release can be installed with
pip install ffq
The development version can be installed with
pip install git+https://github.com/pachterlab/ffq
Usage
Fetch information of an accession and display it in the terminal
ffq [accession]
where [accession] is either:
-
an SRA/EBI/DDJ accession
- (
SRR,SRX,SRSorSRP) - (
ERR,ERX,ERSorERP) - (
DRR,DRS,DRXorDRP)
- (
-
a GEO accession (
GSEorGSM) -
an ENCODE accession (
ENCSR,ENCSBorENCSD) -
a Bioproject accession (
CXR) -
a Biosample accession (
SAMN) -
a DOI
Examples:
$ ffq SRR9990627
#=> Returns metadata for the SRR9990627 run.
$ ffq SRX7347523
#=> Returns metadata for the experiment SRX7347523 and for its associated SRR run.
$ ffq GSE129845
#=> Returns metadata for GSE129845 and for its 5 associated GSM, SRS, SRX and SRR ids.
$ ffq DRP004583
#=> Returns metadata for the study DRP004583 and its 104 associated DRS, DRX and SRR ids.
$ ffq ENCSR998WNE
#=> Returns metadata for the ENCODE experiment ENCSR998WNE.
Fetch information of multiple accessions and display it in the terminal
ffq [accession 1] [accession 2] ...
where [accession 1] and [accession 2] are accessions belonging to any of the above usage example categories.
Examples:
$ ffq SRR11181954 SRR11181954 SRR11181956
#=> Returns metadata for the three SRR runs.
$ ffq GSM4339769 GSM4339770 GSM4339771
#=> Returns metadata for the three GSM accessions, as well as for their corresponding downstream SRS, SRX and SRR accessions.
Fetch information of an accession only down to specified level
ffq -l [level] [accession]
where [level] is the number of downstream accessions you want to fetch
Examples:
$ ffq -l 1 GSM4339769
#=> Returns metadata only for GSM4339769, and not from any downstream accession.
$ ffq -l 3 GSE115469
#=> Returns metadata for GSE115469 and its downstream GSM and SRS accessions.
Fetch only raw data links from the host of your choice and display it in the terminal
FTP host
ffq --ftp [accession(s)]
where [accession(s)] is either a single accession or a space-delimited list of accessions.
AWS host
ffq --aws [accession(s)]
GCP host
ffq --gcp [accession(s)]
NCBI host
ffq --ncbi [accession(s)]
Examples:
# FTP with an SRR
$ ffq --ftp SRR10668798
[
{
"accession": "SRR10668798",
"filename": "SRR10668798_1.fastq.gz",
"filetype": "fastq",
"filesize": 31876537192,
"filenumber": 1,
"md5": "bf8078b5a9cc62b0fee98059f5b87fa7",
"urltype": "ftp",
"url": "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR106/098/SRR10668798/SRR10668798_1.fastq.gz"
},
...
# FTP with a GSE
$ ffq --ftp GSE115469
[
{
"accession": "SRR7276474",
"filename": "P1TLH.bam",
"filetype": "bam",
"filesize": 48545467653,
"filenumber": 1,
"md5": "d0fde6bf21d9f97bdf349a3d6f0a8787",
"urltype": "ftp",
"url": "ftp://ftp.sra.ebi.ac.uk/vol1/SRA716/SRA716608/bam/P1TLH.bam"
},
...
# AWS with SRX
$ ffq --aws SRX7347523
[
{
"accession": "SRR10668798",
"filename": "T84_S1_L001_R1_001.fastq.1",
"filetype": "fastq",
"filesize": null,
"filenumber": 1,
"md5": null,
"urltype": "aws",
"url": "s3://sra-pub-src-6/SRR10668798/T84_S1_L001_R1_001.fastq.1"
},
...
# GCP with ERS
$ ffq --gcp ERS3861775
[
{
"accession": "ERR3585496",
"filename": "4834STDY7002879.bam.1",
"filetype": "bam",
"filesize": null,
"filenumber": 1,
"md5": null,
"urltype": "gcp",
"url": "gs://sra-pub-src-17/ERR3585496/4834STDY7002879.bam.1"
}
]
# NCBI with GSM
$ ffq --ncbi GSM2905292
[
{
"accession": "SRR6425163",
"filename": "SRR6425163.1",
"filetype": "sra",
"filesize": null,
"filenumber": 1,
"md5": null,
"urltype": "ncbi",
"url": "https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos2/sra-pub-run-13/SRR6425163/SRR6425163.1"
}
]
Write accession information to a single JSON file
ffq -o [JSON_PATH] [accession(s)]
where [JSON_PATH] is the path to the JSON file that will contain the information
and [accession(s)] is either a single accession or a space-delimited list of accessions.
Write accession information to multiple JSON files, one file per accession
ffq -o [OUT_DIR] --split [accessions]
where [OUT_DIR] is the path to directory to which to write the JSON files and [accessions] is a space-delimited list of accessions.
Information about each accession will be written to its own separate JSON file named [accession].json.
Fetch information of all studies (and all of their runs) in one or more papers
ffq [DOIS]
where [DOIS] is a space-delimited list of one or more DOIs. The output is a JSON-formatted string (or a JSON file if -o is provided) with SRA study accessions as keys. When --split is also provided, each study is written to its own separate JSON.
Complete output examples
Examples of complete outputs are available in the examples directory.
Downloading data
ffq is specifically designed to download metadata and to facilitate obtaining links to sequence files. To download raw data from the links obtained with ffq you can use one of the following:
cURLandwgetfor FTP links,awsfor AWS links,gsutilfor GCP links,fasterq dumpfor converting SRA files to FASTQ files.
FTP
By default, cURL is installed on most computers and can be used to download files with FTP links. Alternatively, wget can be used.
# Obtain FTP links
$ ffq --ftp SRR10668798
[
{
"accession": "SRR10668798",
"filename": "SRR10668798_1.fastq.gz",
"filetype": "fastq",
"filesize": 31876537192,
"filenumber": 1,
"md5": "bf8078b5a9cc62b0fee98059f5b87fa7",
"urltype": "ftp",
"url": "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR106/098/SRR10668798/SRR10668798_1.fastq.gz"
},
{
"accession": "SRR10668798",
"filename": "SRR10668798_2.fastq.gz",
"filetype": "fastq",
"filesize": 43760586944,
"filenumber": 2,
"md5": "351df47dca211c1f66ef327e280bd4fd",
"urltype": "ftp",
"url": "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR106/098/SRR10668798/SRR10668798_2.fastq.gz"
}
]
# Download the files one-by-one
$ curl -O ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR106/098/SRR10668798/SRR10668798_1.fastq.gz
$ curl -O ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR106/098/SRR10668798/SRR10668798_2.fastq.gz
Alternatively, the urls can be extracted from the json output with jq and then piped into cURL.
$ ffq --ftp SRR10668798 | jq -r '.[] | .url' | xargs curl -O
If you don't have jq installed, you can use the default program grep.
$ ffq --ftp SRR10668798 | grep -Eo '"url": "[^"]*"' | grep -o '"[^"]*"$' | xargs curl -O
AWS
In order to download files from AWS, the aws tool must be installed and credentials must be setup.
# Pipe AWS links to aws s3 cp and download
$ ffq --aws SRX7347523 | jq -r '.[] | .url' | xargs -I {} aws s3 cp {} .
GCP
In order to download files from GCP, the gsutil tool must be install and credentials must be setup.
# Pipe GCP links to gsutil cp and download
$ ffq --gcp ERS3861775 | jq -r '.[] | .url' | xargs -I {} gsutil cp {} .
NCBI-SRA
SRA files downloaded from NCBI can be converted to FASTQ files using fastq-dump or the improved fasterq-dump both of which are installed as part of SRA Toolkit.
# Pipe SRA link to curl and download the SRA file
$ ffq --ncbi GSM2905292 | jq -r '.[] | .url' | xargs curl -O
# Convert the SRA file to FASTQ files with one of the following
$ fastq-dump ./SRR6425163.1 --split-files --include-technical -O ./SRR6425163 --gzip
$ fasterq-dump ./SRR6425163.1 --split-files --include-technical -O ./SRR6425163 # fasterq-dump does not have gzip option
Use cases
ffq facilitates the acquisition of publicly available sequencing data to help answer relevant research questions.
The following was submitted by @sbooeshaghi.
# Goal: quantify publicly available scRNAseq data
$ pip install kb-python gget ffq
$ kb ref -i index.idx -g t2g.txt -f1 transcriptome.fa $(gget ref --ftp -w dna,gtf homo_sapiens)
$ kb count -i index.idx -g t2g.txt -x 10xv3 -o out $(ffq --ftp SRR10668798 | jq -r '.[] | .url' | tr '\n' ' ')
# -> count matrix in out/ folder
# Goal: count the total number of reads
$ ffq SRR10668798 | jq '.. | ."ENA-SPOT-COUNT"? | select(. != null)' | paste -sd+ - | bc
624886427
# Goal: check the total size of the FASTQ files
$ ffq --ftp SRR10668798 | jq '.[] | .filesize ' | paste -sd+ - | bc | numfmt --to=iec-i --suffix=B
71GiB
# Goal: count the number of FASTQ files
$ ffq --ftp SRR10668798 | jq -r 'length'
2
# Goal: get sequence stats for the first 100 entries with seqkit
$ curl -s $(ffq --ftp SRR10668798 | jq -r '.[0] | .url') | zcat | head -400 | seqkit stats -a
file format type num_seqs sum_len min_len avg_len max_len Q1 Q2 Q3 sum_gap N50 Q20(%) Q30(%)
- FASTQ DNA 100 2,600 26 26 26 13 26 13 0 26 95.31 92.92
The following was submitted by @agalvezm.
# Goal: print the first 3 sequences of read 1 to the screen
$ curl -s $(ffq --ftp SRR10668798 | jq -r '.[0] | .url') | zcat | awk '(NR-2)%4==0' | head -n
NCCAAATAGGAATTACATACACCCCC
NAACCTGAGTAGATGTGTTGTTAACT
NGATCTGAGAACTCGGAACTATTTTC
# Goal: get number of counts per unique read sequence from the first 10000 reads
$ curl -s $(ffq --ftp accession | jq -r '.[0] | .url') | zcat | awk '(NR-2)%4==0'| head -n 10000 | sort | uniq -c | sort -r
4 TACACGACACTTAACGATCGGCCTTC
4 GTACTTTAGGCCCGTTTGTGTGCGAT
4 GACGGCTAGTACATGATATAACAAGC
...
The following was submitted by @telatin.
# Goal: concurrent download of a set of FASTQ files given a list of IDs (list.txt)
# (Requires Nextflow and Docker, or Conda, to be installed. Pipeline and dependencies will be installed automatically)
$ nextflow run telatin/getreads -r main -profile docker --list list.txt --outdir downloaded-reads/
For instructions on how to install Nextflow and Docker, or Conda, see the installation instructions.
Do you have a cool use case for ffq? Submit a PR (including the goal, code snippet, and your username) so that we can feature it here.
Failure modes
Many factors, independent of ffq, may result in failure to fetch metadata or missing metadata including:
- broken internet connection
- improperly formatted accession
- recently submitted data to SRA (not synced with ENA)
- exceeded request rate for servers
- missing metadata from online database
If you believe you have identified a bug in ffq please see the section on contributing*.
Contributing
Thank you for wanting to improve ffq! If you have a bug that is related to ffq please create an issue. The issue should contain
- the
ffqcommand ran with--verbose, - the error message, and
- the
ffqandpythonversion.
Please make all Pull Requests against the devel branch and include a message detailing the exact changes made, the reasons for the change, and tests that check for the correctness of those changes.
Some tips for improving the ffq code base:
- the developer dependencies can be installed with
pip install -r dev-requirements.txt - unit tests can be added to the
./tests/test_*.py - code reformatting can be performed by running
black ffq/ - code quality can be checked by running
make check - tests can be performed by running
make test
Caveats and limitations
ffq relies on the information provided by the different APIs it uses to retrieve metadata (hosted by ENA, NCBI, ENCODE, etc). Therefore, returning consistent and accurate metadata is dependent on the accuracy and consistency of such databases. Unfortunately, we have observed instances where some APIs are updated without notice. This leads to unconsistent metadata retrieval by ffq that cannot be solved on our end.
For example, as of May 29th, the command:
ffq --ncbi SRR6835844
returned:
[{'accession': 'SRR6835844',
'filename': 'SRR6835844.1',
'filenumber': 1,
'filesize': None,
'filetype': 'sra',
'md5': None,
'url': 'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos2/sra-pub-run-13/SRR6835844/SRR6835844.1',
'urltype': 'ncbi'}]
On June 1st, we detected an error in one of ffq’s tests. Running the same command led to the following output:
[]
Investigating this issue, we discovered that the output of the eutil’s efetch tool had changed (for a comparison, compare files SRR6835844_altlinks_old.txt and SRR6835844_altlinks_new.txt contained in tests/fixtures). In the new output, ncbi hosted links were no longer provided. This affects a large number of accessions, not only SRR6835844. We have updated our tests accordingly and will continue to monitor the situation.
Naming
ffq is short for FetchFastQ.
Cite
@article{galvez2022metadata,
title={Metadata retrieval from sequence databases with ffq},
author={G{\'a}lvez-Merch{\'a}n, {\'A}ngel and Min, Kyung Hoi Joseph and Pachter, Lior and Booeshaghi, A. Sina},
year={2022}
}
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ffq-0.3.1.tar.gz.
File metadata
- Download URL: ffq-0.3.1.tar.gz
- Upload date:
- Size: 32.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
02a82b1130fa1d50558b8e3b9663cc2db1ad95498a7345783e69517f6391884b
|
|
| MD5 |
4b647cb18939ddfc7faff56a7199acbb
|
|
| BLAKE2b-256 |
41a624a1edbf55edc72d3451152078235bf27aa59628fd0d035d0ffee5ccb425
|
File details
Details for the file ffq-0.3.1-py3-none-any.whl.
File metadata
- Download URL: ffq-0.3.1-py3-none-any.whl
- Upload date:
- Size: 25.7 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
064ef79fc1bf187029a2352794d5c74cc4235718c3778466dda34b5efb6981b7
|
|
| MD5 |
4fd005f7cce5d7d6ab72714cfa97d53d
|
|
| BLAKE2b-256 |
3b383664622423e18857a7a610d6b532a6eaa0684b2a4b97b9fe75bfaf95d108
|







