Single Cell GeneVector Library

Project description
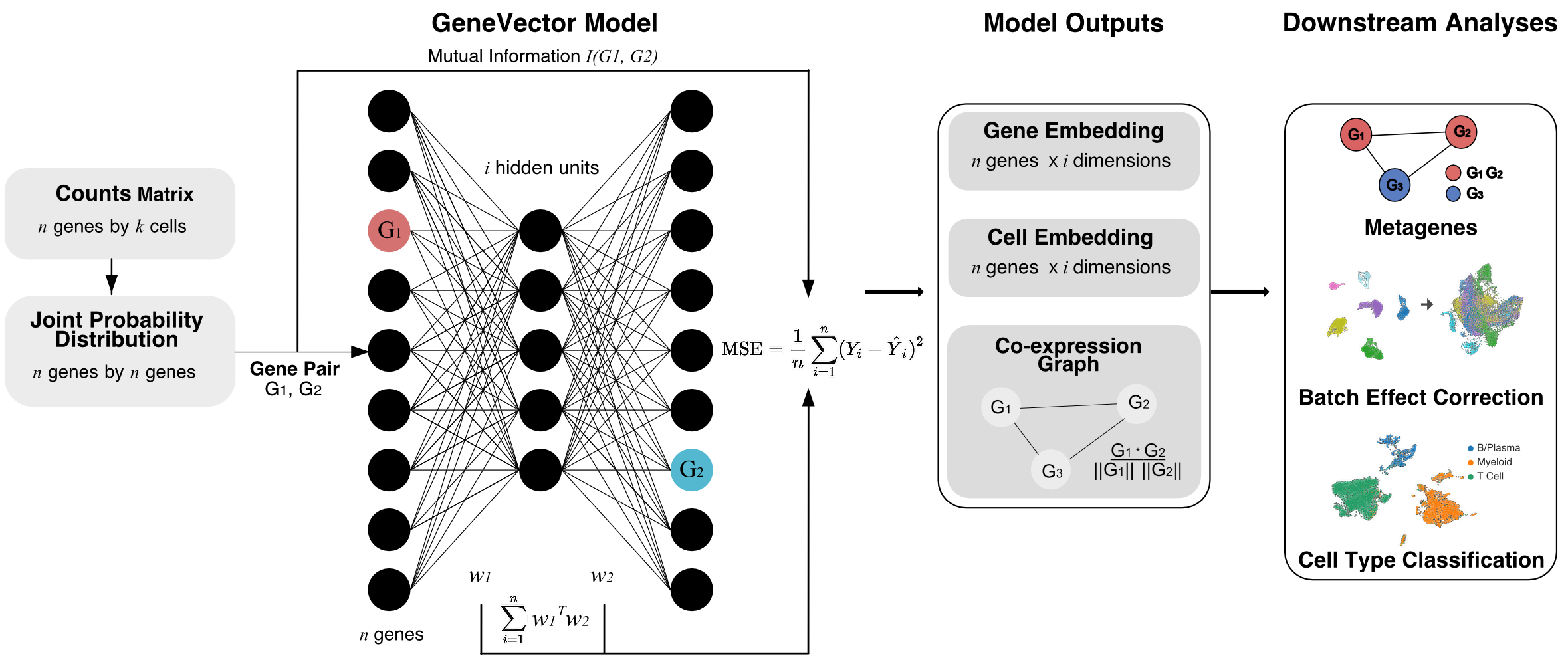
GeneVector





GeneVector is a Python library for single-cell RNA sequencing analysis that learns distributed gene representations using a neural embedding approach. It enables gene co-expression analysis, cell type annotation, and metagene discovery through vector arithmetic operations.
Key Features
- Gene Embeddings: Learn distributed representations of genes based on co-expression patterns
- Cell Type Annotation: Automated cell type assignment using marker gene sets
- Metagene Discovery: Identify functionally related gene modules through clustering
- Vector Arithmetic: Perform gene relationship analysis using vector operations
- Batch Correction: Fast batch effect correction for multi-sample datasets
- Pluggable Targets: Train on mutual information, Pearson, Spearman, Jaccard, cosine, or custom metrics
- High Performance: Numba JIT, Rust native, and CUDA GPU backends for MI computation
- Smart Caching: Computed scores cached to disk, instant reload on re-runs
Installation
From PyPI (Recommended)
pip install genevector
With Numba Acceleration (Recommended)
pip install genevector[fast]
From Source
git clone https://github.com/nceglia/genevector.git
cd genevector
pip install -e .
# with numba support
pip install -e ".[fast]"
# with rust backend (requires Rust toolchain)
pip install maturin
maturin develop --release
Dependencies
- Python ≥ 3.9
- PyTorch
- Scanpy
- NumPy, SciPy, Pandas
- Matplotlib, Seaborn
Optional: numba for JIT-accelerated MI computation (strongly recommended).
Quick Start
import scanpy as sc
from genevector.data import GeneVectorDataset
from genevector.model import GeneVector
from genevector.embedding import GeneEmbedding, CellEmbedding
# Load your single-cell data
adata = sc.read_h5ad("your_data.h5ad")
# Create dataset (auto-selects fastest MI backend)
dataset = GeneVectorDataset(adata)
# Train the model
model = GeneVector(dataset, output_file="genes.vec", emb_dimension=100)
model.train(1000, threshold=1e-6)
# Load gene embeddings
gene_embed = GeneEmbedding("genes.vec", dataset, vector="average")
# Generate cell embeddings
cell_embed = CellEmbedding(dataset, gene_embed)
adata_embedded = cell_embed.get_adata()
# Analyze gene similarities
similarities = gene_embed.compute_similarities("CD8A")
gene_embed.plot_similarities("CD8A", n_genes=10)
MI Computation Backends
GeneVector computes pairwise co-expression scores between all genes as the training target. For mutual information (the default target), multiple backends are available with automatic dispatch.
Dispatch Priority
When mi_backend="auto" (the default), GeneVector selects the fastest available backend:
| Priority | Backend | Speedup vs Legacy | How It Works | Availability |
|---|---|---|---|---|
| 1 | CUDA GPU | ~200-1000x | PyTorch scatter-based histograms on GPU | Requires device="cuda" and NVIDIA GPU |
| 2 | Rust | ~100-800x | Native compiled with rayon parallelism | Requires maturin develop --release build |
| 3 | Numba | ~100-500x | JIT-compiled, parallel across CPU cores | Requires pip install numba |
| 4 | NumPy | ~10-30x | Vectorized discretization, Python pair loop | Always available (default fallback) |
Selecting a Backend
# Auto-select (recommended): picks the fastest available
dataset = GeneVectorDataset(adata, mi_backend="auto")
# Force a specific backend
dataset = GeneVectorDataset(adata, mi_backend="rust")
dataset = GeneVectorDataset(adata, mi_backend="numba")
dataset = GeneVectorDataset(adata, mi_backend="numpy")
dataset = GeneVectorDataset(adata, mi_backend="gpu", device="cuda")
Check Available Backends
from genevector.metrics import HAS_NUMBA
# Rust availability (if built)
try:
from genevector._rust import compute_mi_pairs
print("Rust backend: available")
except ImportError:
print("Rust backend: not installed")
print(f"Numba backend: {'available' if HAS_NUMBA else 'not installed'}")
import torch
print(f"CUDA backend: {'available' if torch.cuda.is_available() else 'not available'}")
Practical Runtimes (Approximate)
For 2,000 genes × 5,000 cells (~2M gene pairs):
| Backend | Time |
|---|---|
| Legacy (v0.2) | ~26 hours |
| NumPy | ~3 hours |
| Numba | ~2-3 minutes |
| Rust | ~1-2 minutes |
Scores are cached to ~/.genevector/cache/ after the first run. Subsequent runs with the same data load instantly.
Co-expression Targets
GeneVector can train on different co-expression metrics beyond mutual information:
# Default: signed mutual information
dataset = GeneVectorDataset(adata, target="mi", signed_mi=True)
# Pearson correlation
dataset = GeneVectorDataset(adata, target="pearson")
# Spearman rank correlation
dataset = GeneVectorDataset(adata, target="spearman")
# Jaccard index (binarized co-detection)
dataset = GeneVectorDataset(adata, target="jaccard")
# Cosine similarity between gene expression vectors
dataset = GeneVectorDataset(adata, target="cosine")
# Custom callable
def my_metric(X, gene_names, **kwargs):
# must return dict[str, dict[str, float]]
...
dataset = GeneVectorDataset(adata, target=my_metric)
The mi_backend parameter only applies when target="mi". The matrix-based targets (Pearson, Spearman, Jaccard, cosine) compute in seconds via BLAS regardless of gene count.
Tutorials
See the example/ directory for comprehensive workflows:
- PBMC workflow: Identification of interferon stimulated metagene and cell type annotation
- TICA workflow: Cell type assignment
- SPECTRUM workflow: Vector arithmetic for site specific metagenes
- FITNESS workflow: Identifying increasing metagenes in time series
Detailed Usage Guide
1. Data Loading
GeneVector uses Scanpy AnnData objects and requires raw count data in the .X matrix. It's recommended to subset genes using the seurat_v3 flavor in Scanpy or GeneVector's entropy-based quality control.
from genevector.data import GeneVectorDataset
from genevector.model import GeneVector
from genevector.embedding import GeneEmbedding, CellEmbedding
import scanpy as sc
# Load and preprocess data
adata = sc.read_h5ad("your_data.h5ad")
# Option A: Scanpy's variable gene selection
sc.pp.highly_variable_genes(adata, flavor="seurat_v3", n_top_genes=2000)
adata = adata[:, adata.var.highly_variable]
# Option B: GeneVector's entropy-based gene filtering
adata = GeneVectorDataset.quality_control(adata, entropy_threshold=1.0)
# Create dataset
dataset = GeneVectorDataset(adata)
2. Model Training
Creating a GeneVector object computes the co-expression target (MI by default) and prepares training batches. Training time varies by dataset size — a 10k PBMC dataset trains in under 5 minutes. The emb_dimension parameter controls vector size (minimum 50 recommended).
# Initialize model (triggers MI computation + batch preparation)
model = GeneVector(
dataset,
output_file="genes.vec",
emb_dimension=100,
device="cpu"
)
# Train for 1000 iterations or until convergence
model.train(1000, threshold=1e-6)
# Visualize training progress
model.plot()
3. Gene Embeddings Analysis
Training produces two vector files (input and output weights). Using the average of both weights is recommended for best results.
# Load gene embeddings
gene_embed = GeneEmbedding("genes.vec", dataset, vector="average")
# Compute gene similarities
similarities_df = gene_embed.compute_similarities("CD8A")
gene_embed.plot_similarities("CD8A", n_genes=10)
# Generate metagenes through clustering
gene_adata = gene_embed.get_adata(resolution=40)
metagenes = gene_embed.get_metagenes(gene_adata)
# Visualize specific metagenes
gene_embed.plot_metagene(gene_adata, mg=metagenes[0])
4. Cell Embeddings
Generate cell embeddings using the trained gene vectors. The embeddings are stored in AnnData format with automatic UMAP generation.
# Create cell embeddings
cell_embed = CellEmbedding(dataset, gene_embed)
adata_embedded = cell_embed.get_adata()
# Visualize with Scanpy
sc.pl.umap(adata_embedded)
# Optional: Batch correction
cell_embed.batch_correct(column="sample", reference="control")
adata_corrected = cell_embed.get_adata()
5. Cell Type Assignment
# Define marker genes for cell types
markers = {
"T Cell": ["CD3D", "CD3G", "CD3E", "TRAC", "IL32", "CD2"],
"B/Plasma": ["CD79A", "CD79B", "MZB1", "CD19", "BANK1"],
"Myeloid": ["LYZ", "CST3", "AIF1", "CD68", "C1QA", "C1QB", "C1QC"]
}
# Perform automated cell type assignment
annotated_adata = cell_embed.phenotype_probability(adata_embedded, markers)
# Visualize results
prob_cols = [col for col in annotated_adata.obs.columns if "Pseudo-probability" in col]
sc.pl.umap(annotated_adata, color=prob_cols + ["genevector"], size=25)
6. Caching
Computed co-expression scores are cached automatically. Control this with:
# Enable caching (default)
dataset = GeneVectorDataset(adata, use_cache=True)
# Disable caching (always recompute)
dataset = GeneVectorDataset(adata, use_cache=False)
# Clear the cache
from genevector.cache import clear_cache
clear_cache()
Cache location: ~/.genevector/cache/. Cache keys incorporate the expression matrix, gene list, target function, and all parameters, so different configurations never collide.
Contributing
Contributions are welcome! Please feel free to submit a Pull Request. For major changes, please open an issue first to discuss what you would like to change.
License
This project is licensed under the MIT License - see the LICENSE file for details.
Citation
If you use GeneVector in your research, please cite:
Ceglia, N., Sethna, Z., Freeman, S.S. et al. Identification of transcriptional programs using dense vector representations defined by mutual information with GeneVector. Nat Commun 14, 4400 (2023). https://doi.org/10.1038/s41467-023-39985-2
@article{ceglia2023genevector,
title={Identification of transcriptional programs using dense vector representations defined by mutual information with GeneVector},
author={Ceglia, Nicholas and Sethna, Zachary and Freeman, Samuel S and others},
journal={Nature Communications},
volume={14},
pages={4400},
year={2023},
doi={10.1038/s41467-023-39985-2}
}
Documentation
For detailed documentation and examples, visit: https://genevector.readthedocs.io


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distributions
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file genevector-1.0.0.tar.gz.
File metadata
- Download URL: genevector-1.0.0.tar.gz
- Upload date:
- Size: 54.4 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: maturin/1.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4a7c66a34a0f712ef32556cd54f0b65d76ee0e573bf69b7405645312848552a9
|
|
| MD5 |
81c45bef58014a17f551c8f46cd4106d
|
|
| BLAKE2b-256 |
fa6b81c8879b15a3e2ac82793e1338868b5c520372e72810830aeb7be99dba83
|
File details
Details for the file genevector-1.0.0-cp311-cp311-macosx_11_0_arm64.whl.
File metadata
- Download URL: genevector-1.0.0-cp311-cp311-macosx_11_0_arm64.whl
- Upload date:
- Size: 306.3 kB
- Tags: CPython 3.11, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? No
- Uploaded via: maturin/1.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
35c746f1b331b87bee498fa673b08eaa5df65b440601b8a4ee4c12a7b2420941
|
|
| MD5 |
f95f1afba72614ee73ab0eb7242f5299
|
|
| BLAKE2b-256 |
c1cc834418a188f903ca77fbb451daba92429553597ffeac132be5b7e32543d0
|
File details
Details for the file genevector-1.0.0-cp311-cp311-macosx_10_12_x86_64.whl.
File metadata
- Download URL: genevector-1.0.0-cp311-cp311-macosx_10_12_x86_64.whl
- Upload date:
- Size: 312.5 kB
- Tags: CPython 3.11, macOS 10.12+ x86-64
- Uploaded using Trusted Publishing? No
- Uploaded via: maturin/1.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c9044ba244cc0e3dc68f115cb2b00a033c7c3acbde48359b9d860dbeac0d7b59
|
|
| MD5 |
3de02922cfcb89f95bfb12161f8c13e9
|
|
| BLAKE2b-256 |
0737a9a46a71b0631da70805e4f2c0dda5218db01a65f6c52cf22fd37849658c
|
File details
Details for the file genevector-1.0.0-cp38-cp38-manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: genevector-1.0.0-cp38-cp38-manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 361.0 kB
- Tags: CPython 3.8, manylinux: glibc 2.17+ x86-64
- Uploaded using Trusted Publishing? No
- Uploaded via: maturin/1.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f4e56235865522bdc1c42376ef853bf1f5a943334801c142efb969d330ba462f
|
|
| MD5 |
9a7c43970620fca4f8bf69f079ec2a73
|
|
| BLAKE2b-256 |
fa7ed990c704f7b33c2304208aac9ceee43f8960c0f47fa2e197bd37d741c533
|







