Python scripts to find enrichment of GO terms

Project description
GOATOOLS: A Python library for Gene Ontology analyses




| Authors | Haibao Tang (tanghaibao) |
| DV Klopfenstein (dvklopfenstein) | |
| Brent Pedersen (brentp) | |
| Fidel Ramirez (fidelram) | |
| Aurelien Naldi (aurelien-naldi) | |
| Patrick Flick (patflick) | |
| Jeff Yunes (yunesj) | |
| Kenta Sato (bicycle1885) | |
| Chris Mungall (cmungall) | |
| Greg Stupp (stuppie) | |
| David DeTomaso (deto) | |
| Olga Botvinnik (olgabot) | |
| tanghaibao@gmail.com | |
| License | BSD |
How to cite
[!TIP] GOATOOLS is now published in Scientific Reports!
Klopfenstein DV, ... Tang H (2018) GOATOOLS: A Python library for Gene Ontology analyses Scientific reports
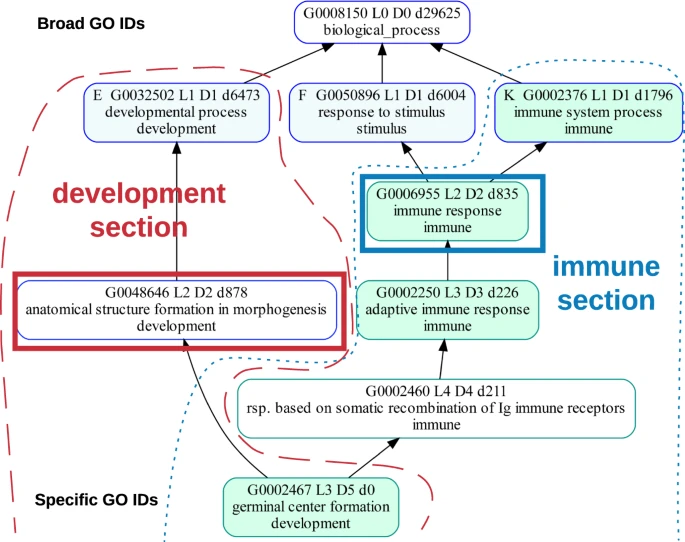
- GO Grouping: Visualize the major findings in a gene ontology enrichment analysis (GOEA) more easily with grouping. A detailed description of GOATOOLS GO grouping is found in the manuscript.
- Compare GO lists:
Compare two or more lists
of GO IDs using
goatools compare_gos, which can be used with or without grouping. - Stochastic GOEA simulations: One of the findings resulting from our simulations is: Larger study sizes result in higher GOEA sensitivity, meaning fewer truly significant observations go unreported. The code for the stochastic GOEA simulations described in the paper is found here
Contents
This package contains a Python library to
-
Process over- and under-representation of certain GO terms, based on Fisher's exact test. With numerous multiple correction routines including locally implemented routines for Bonferroni, Sidak, Holm, and false discovery rate. Also included are multiple test corrections from statsmodels: FDR Benjamini/Hochberg, FDR Benjamini/Yekutieli, Holm-Sidak, Simes-Hochberg, Hommel, FDR 2-stage Benjamini-Hochberg, FDR 2-stage Benjamini-Krieger-Yekutieli, FDR adaptive Gavrilov-Benjamini-Sarkar, Bonferroni, Sidak, and Holm.
-
Process the obo-formatted file from Gene Ontology website. The data structure is a directed acyclic graph (DAG) that allows easy traversal from leaf to root.
-
Read GO Association files:
- GAF (GO Annotation File)
- GPAD (Gene Product Association Data)
- NCBI's gene2go file
- id2gos format. See example
-
Print decendants count and/or information content for a list of GO terms
-
Get parents or ancestors for a GO term with or without optional relationships, including Print details about a GO ID's parents
-
Get children or descendants for a GO term with or without optional relationships
-
Compare two or more lists of GO IDs
-
Group GO terms for easier viewing
-
Map GO terms (or protein products with multiple associations to GO terms) to GOslim terms (analog to the map2slim.pl script supplied by geneontology.org)
Installation
Make sure your Python version >= 3.7, and download an
.obo file of the most current
GO:
wget http://current.geneontology.org/ontology/go-basic.obo
or .obo file for the most current GO
Slim terms (e.g.
generic GOslim) :
wget http://current.geneontology.org/ontology/subsets/goslim_generic.obo
PyPI
pip install goatools
To install the development version:
pip install git+git://github.com/tanghaibao/goatools.git
Bioconda
conda install -c bioconda goatools
Dependencies
When installing via PyPI or Bioconda as described above, all dependencies are automatically downloaded. Alternatively, you can manually install:
-
For statistical testing of GO enrichment:
scipy.stats.fisher_exactstatsmodels(optional) for access to a variety of statistical tests for GOEA
-
To plot the ontology lineage, install one of these two options:
- Graphviz, for graph visualization.
- pygraphviz, Python binding for communicating with Graphviz:
- pydot, a Python interface to Graphviz's Dot language.
Cookbook
run.sh contains example cases using the installed goatools CLI.
Find GO enrichment of genes under study
See examples in find_enrichment
The goatools find_enrichment command takes as arguments files
containing:
- gene names in a study
- gene names in population (or other study if
--compareis specified) - an association file that maps a gene name to a GO category.
Please look at tests/data folder to see examples on how to make these
files. when ready, the command looks like:
goatools find_enrichment --pval=0.05 --indent data/study \
data/population data/association
and can filter on the significance of (e)nrichment or (p)urification. it can report various multiple testing corrected p-values as well as the false discovery rate.
The e in the "Enrichment" column means "enriched" - the concentration
of GO term in the study group is significantly higher than those in
the population. The "p" stands for "purified" - significantly lower
concentration of the GO term in the study group than in the population.
Important note: by default, goatools find_enrichment propagates counts
to all the parents of a GO term. As a result, users may find terms in
the output that are not present in their association file. Use
--no_propagate_counts to disable this behavior.
Write GO hierarchy
goatools wr_hier: Given a GO ID, write the hierarchy below (default) or above (--up) the given GO.
Plot GO lineage
goatools go_plot:- Plots user-specified GO term(s) up to root
- Multiple user-specified GOs
- User-defined colors
- Plot relationships (
-r) - Optionally plot children of user-specfied GO terms
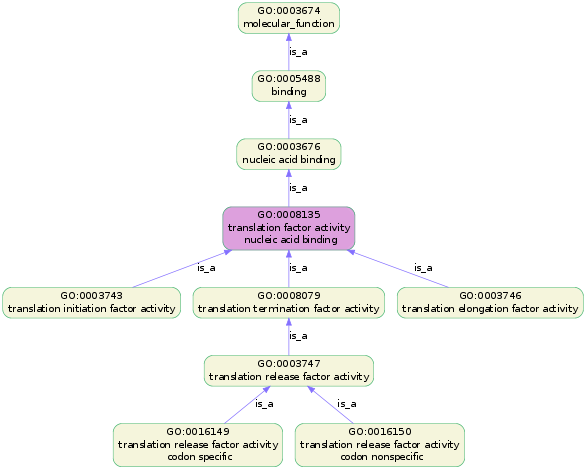
goatools plot_go_termcan plot the lineage of a certain GO term, by:
goatools plot_go_term --term=GO:0008135
This command will plot the following image.
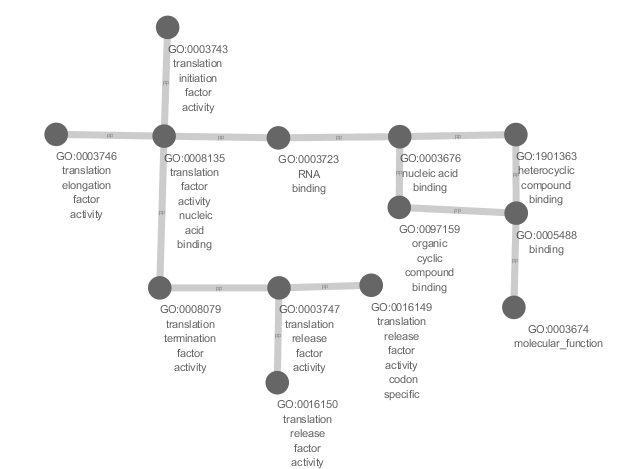
Sometimes people like to stylize the graph themselves, use option
--gml to generate a GML output which can then be used in an external
graph editing software like Cytoscape. The
following image is produced by importing the GML file into Cytoscape
using yFile orthogonal layout and solid VizMapping. Note that the GML
reader plugin may need to be
downloaded and installed in the plugins folder of Cytoscape:
goatools plot_go_term --term=GO:0008135 --gml
Map GO terms to GOslim terms
See goatools map_to_slim for usage. As arguments it takes the gene ontology
files:
- the current gene ontology file
go-basic.obo - the GOslim file to be used (e.g.
goslim_generic.oboor any other GOslim file)
The script either maps one GO term to its GOslim terms, or protein products with multiple associations to all its GOslim terms.
To determine the GOslim terms for a single GO term, you can use the following command:
goatools map_to_slim --term=GO:0008135 go-basic.obo goslim_generic.obo
To determine the GOslim terms for protein products with multiple associations:
goatools map_to_slim --association_file=data/association go-basic.obo goslim_generic.obo
Where the association file has the same format as used for
goatools find_enrichment.
The implementation is similar to map2slim.
Technical notes
Available statistical tests for calculating uncorrected p-values
For calculating uncorrected p-values, we use SciPy:
Available multiple test corrections
We have implemented several significance tests:
bonferroni, bonferroni correctionsidak, sidak correctionholm, hold correctionfdr, false discovery rate (fdr) implementation using resampling
Additional methods are available if statsmodels is installed:
sm_bonferroni, bonferroni one-step correctionsm_sidak, sidak one-step correctionsm_holm-sidak, holm-sidak step-down method using Sidak adjustmentssm_holm, holm step-down method using Bonferroni adjustmentssimes-hochberg, simes-hochberg step-up method (independent)hommel, hommel closed method based on Simes tests (non-negative)fdr_bh, fdr correction with Benjamini/Hochberg (non-negative)fdr_by, fdr correction with Benjamini/Yekutieli (negative)fdr_tsbh, two stage fdr correction (non-negative)fdr_tsbky, two stage fdr correction (non-negative)fdr_gbs, fdr adaptive Gavrilov-Benjamini-Sarkar
In total 15 tests are available, which can be selected using option
--method. Please note that the default FDR (fdr) uses a resampling
strategy which may lead to slightly different q-values between runs.
iPython Notebooks
Optional attributes
Run a Ontology Enrichment Analysis (GOEA)
goea_nbt3102 human phenotype ontologies
Show many study genes are associated with RNA, translation, mitochondria, and ribosomal
Report level and depth counts of a set of GO terms
Find all human protein-coding genes associated with cell cycle
Calculate annotation coverage of GO terms on various species
Determine the semantic similarities between GO terms
semantic_similarity semantic_similarity_wang
Obsolete GO terms are loaded upon request
Want to Help?
Prior to submitting your pull request, please add a test which verifies your code, and run:
make test
Items that we know we need include:
-
Add code coverage runs
-
Edit tests in the
makefileunder the comment -
Help setting up documentation. We are using Sphinx and Python docstrings to create documentation. For documentation practice, use make targets:
make mkdocs_practiceTo remove practice documentation:
make rmdocs_practiceOnce you are happy with the documentation do:
make gh-pages
Star History

Copyright (C) 2010-2021, Haibao Tang et al. All rights reserved.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file goatools-1.6.5.tar.gz.
File metadata
- Download URL: goatools-1.6.5.tar.gz
- Upload date:
- Size: 17.8 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
0d799706dc3ae4480feda25f411f8e9b2741c0d8ea7ad73af0b05730198a4be1
|
|
| MD5 |
212877b8285264bcd49d8d9b0461cb68
|
|
| BLAKE2b-256 |
e68707e80f31af72d8e2c35936d1e5670ac05ff6009cc4a9412eb99e18efd24a
|
File details
Details for the file goatools-1.6.5-py3-none-any.whl.
File metadata
- Download URL: goatools-1.6.5-py3-none-any.whl
- Upload date:
- Size: 15.8 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
2b093ded287178b494e28350bce6beb60b920ff5f923fc5aaef227dd6ffb935a
|
|
| MD5 |
e2bfc3b3b897dc2ba3d670b14a398a36
|
|
| BLAKE2b-256 |
20531020a7b32651289a68ec853d43fb412c8982d6d1de2289e6cb6f55bc4b7b
|







