Python library for Gene Ontology Reverse Lookup


Project description
GOReverseLookup




GOReverseLookup is a Python package designed for Gene Ontology Reverse Lookup. It serves the purpose of identifying statistically significant genes within a set or a cross-section of selected Gene Ontology Terms. Researchers need only define their own states of interest (SOIs), and select GO terms must be attributed as either positive or negative regulators of the chosen SOIs. For more information regarding the creation of the input file for the program, refer to the Input file section. Once the input file is created, the GOReverseLookup program can be started. Once the algorithm is completed, the program saves statistically significant genes in a standalone file.
For example, if researchers were interested in the angiogenesis SOI, then an attributed group of GO terms as positive regulators of angiogenesis might have been defined using the following GO terms:
- GO:1903672 positive regulation of sprouting angiogenesis
- GO:0001570 vasculogenesis
- GO:0035476 angioblast cell migration
And negative regulators of the angiogenesis SOI might have been defined as the following group:
- GO:1903671 negative regulation of sprouting angiogenesis
- GO:1905554 negative regulation of vessel branching
- GO:0043537 negative regulation of blood vessel endothelial cell migration
If a researcher defines the target process as positive regulation of a desired SOI (in our case angiogenesis), then GOReverseLookup finds all genes statistically relevant for the group of GO terms defined as positive regulators of angiogenesis (p < 0.05) while excluding any genes determined to be statistically significant (p < 0.05) in the opposing process (in our case, negative regulation of angiogenesis). P-value threshold can also be manually set by the user.
Getting Started
This section instructs you how to install the GOReverseLookup package and its prerequisites.
Folder setup
You MUST create a local folder anywhere on your disk, which will be used as the GOReverseLookup's working environment, as well as unified storage for all of your research projects. We advise you to create a folder structure with a folder named goreverselookup as the parent folder (this folder will be used as a local installation location for the GOReverseLookup program), and a subfolder named research_models, where you will store the input files for GOReverseLookup and their results. Therefore, the folder structure should be the following:
.../goreverselookup/
- research_models/
Installation
Python installation
For your computer to understand the GOReverseLookup program, it requires the Python programming language, which MUST be installed. Our program is currently tested on Python versions 3.10.x through 3.11.x, but not yet on 3.12.x. Thus, we advise you to use the Python version 3.11.5, which is available for download from this website. Following this link, navigate to the Files section:
- if you are using Windows: download Windows installer (64-bit)
- if you are using macOS: download macOS 64-bit universal2 installer
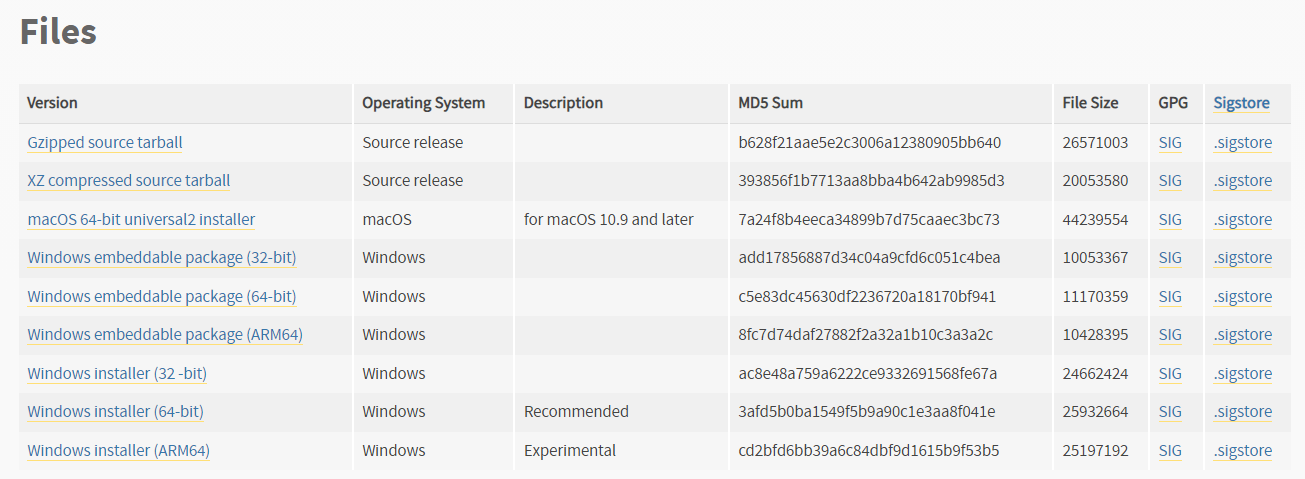
Open the File Explorer program, then open the Downloads folder and run the installer by double clicking it.
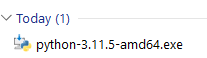
The default Python installer window pops up:
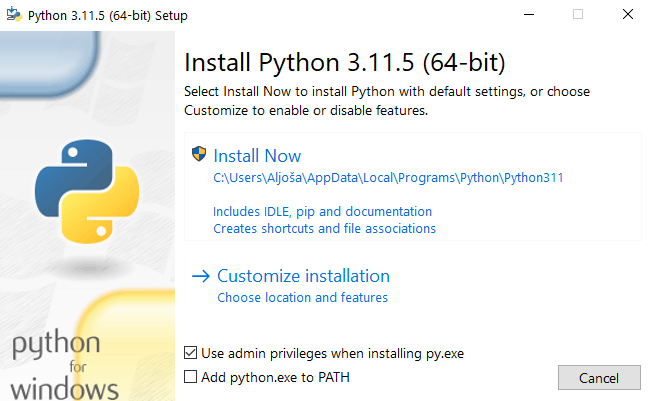
Make sure to also select Add python.exe to PATH. This will make Python available across all-file locations, which is of extreme importance for running Python commands from the console (Command prompt in Windows). Then, click on Install Now. A further observation of the installer's window also reveals that this installer is bundled with PIP (Python's package manager), thus manual installation of PIP won't be necessary. This is important, since PIP will be used to download GOReverseLookup.
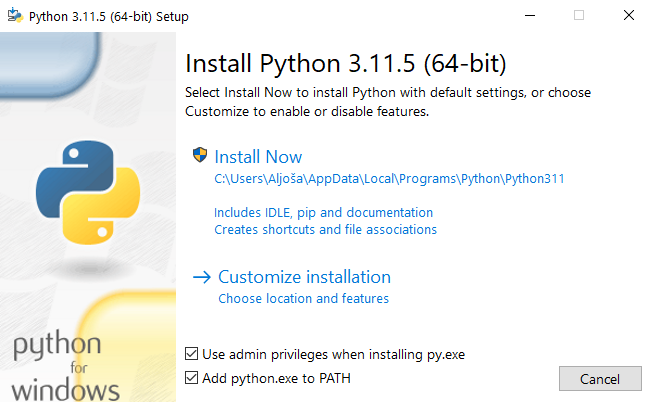
Wait for the installation of Python to finish. Once it is finished, close the installer window.
If you wish to download a specific Python version, browse through the Python's downloads page - for beginners, we advise you to find a release with an available installer.
Then, open the command prompt using the Windows search bar:
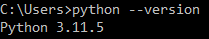
Inside the command prompt, execute the command python --version. If Python installation has been completed successfully, a version of the Python programming language will be displayed:
Also verify that PIP (Python's package manager) is installed. In our instance, it has been mentioned in the Python installer's window that PIP will also be installed along with Python. To verify the installation of PIP, run the pip --version command:

Creating your GOReverseLookup workspace
To create a standalone GOReverseLookup workspace that will be central both to GOReverseLookup's installation files and the research files, create the folder setup as instructed in Folder setup. Create a Python's virtual environment in the goreverselookup folder using the command python -m venv "PATH_TO_GOREVERSELOOKUP". For example, on my computer, the goreverselookup folder exists at F:\Development\python_environments\goreverselookup, thus the command to create the virtual environment is: python -m venv "F:\Development\python_environments\goreverselookup":

To find the path to your goreverselookup folder, open the goreverselookup folder in the File Explorer and click on the Address Bar, then copy the filepath.


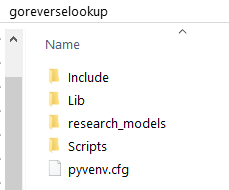
After running the virtual environment creation command, you should notice the goreverselookup folder be populated with new folders: Include, Lib and Scripts, and a file named pyvenv.cfg. These belong to the newly created Python's virtual environment, so do not change their contents in any way. As stated in the Folder setup section, the goreverselookup folder also contains a research_models folder.
To activate the newly created virtual environment, there exists an activation script named activate.bat in the newly created Scripts folder. You will need to activate this virtual environment in command prompt every time you begin working with GOReverseLookup, thus we advise you to save the activation command in a text file somewhere easily accessible, such as your desktop. To activate the virtual environment, just supply the path to the activation script to the command prompt - in our case, the path to the activation script is F:\Development\python_environments\goreverselookup\Scripts\activate. After running this in command prompt, the virtual environment will be activated:
Installing GOReverseLookup
As per instructions in Creating your GOReverseLookup workspace, activate the newly created virtual environment, so the current command prompt pointer points to the virtual environment. E.g.:
Now, run the command pip install goreverselookup and wait for the installation to complete:

To confirm the installation, run the command pip list and find the goreverselookup package, along with it's version:
Usage
Creating the input file
The entry to the program is an input file, which is ideally placed in the .../goreverselookup/research_models/ folder, as explained in Folder setup. It contains all the relevant data for the program to complete the analysis of statistically important genes that positively or negatively contribute to one or more states of interest.
An example input.txt file to discover the genes that positively contribute to both the development of chronic inflammation and cancer is supplied below.
WARNING: This is just an example input file intended to give a quick overview of the general contents of an input file. It is not meant to be used in an analysis, as it contains far too few GO terms to discover any potential genes. To test if your program setup works, use research-grade input files, which are located in the research_models folder, e.g. research_models/chronic-inflammation_cancer/IEA+/ind_ann,p=0.05,IEA+ (145)/input.txt.
# Comments are preceded by a single '#'. Comment lines will not be parsed in code.
# Section titles are preceded by three '###'
# The values at each line are usually delineated using the TAB character. E.g. pvalue 0.05 (pvalue and it's value 0.05 are separated by a TAB).
#
###evidence_code_groups
experimental EXP_ECO:0000269,IDA_ECO:0000314,IPI_ECO:0000353,IMP_ECO:0000315,IGI_ECO:0000316,IEP_ECO:0000270,HTP_ECO:0006056,HDA_ECO:0007005,HMP_ECO:0007001,HGI_ECO:0007003,HEP_ECO:0007007
phylogenetic IBA_ECO:0000318,IBD_ECO:0000319,IKR_ECO:0000320,IRD_ECO:0000321
computational_analysis ISS_ECO:0000250,ISO_ECO:0000266,ISA_ECO:0000247,ISM_ECO:0000255,IGC_ECO:0000317,RCA_ECO:0000245
author_statement TAS_ECO:0000304,NAS_ECO:0000303
curator_statement IC_ECO:0000305,ND_ECO:0000307
electronic IEA_ECO:0000501
###settings
pvalue 0.05
multiple_correction_method fdr_bh
target_organism homo_sapiens|UniProtKB|NCBITaxon:9606 # format: organism_label|organism_database|ncbi_taxon
ortholog_organisms danio_rerio|ZFIN|NCBITaxon:7955,rattus_norvegicus|RGD|NCBITaxon:10116,mus_musculus|MGI|NCBITaxon:10090,xenopus_tropicalis|Xenbase|NCBITaxon:8364
evidence_codes experimental(~),phylogenetic(~),computational_analysis(~),author_statement(TAS),!curator_statement(ND),!electronic(~)
#evidence_codes experimental(~),phylogenetic(~),computational_analysis(~),author_statement(TAS),!curator_statement(ND),electronic(~)
gorth_ortholog_fetch_for_indefinitive_orthologs True
gorth_ortholog_refetch False
fisher_test_use_online_query False
include_indirect_annotations False p
indirect_annotations_max_depth -1
uniprotkb_genename_online_query False
goterm_gene_query_timeout 240
goterm_gene_query_max_retries 3
exclude_opposite_regulation_direction_check False
goterm_name_fetch_async True
goterm_gene_fetch_async True
goterm_name_fetch_req_delay 1.5
goterm_name_fetch_max_connections 5
goterm_gene_fetch_req_delay 0.8
goterm_gene_fetch_max_connections 5
###filepaths
go_obo data_files/go.obo https://purl.obolibrary.org/obo/go.obo all
goa_human data_files/goa_human.gaf http://geneontology.org/gene-associations/goa_human.gaf.gz homo_sapiens
#goa_zfin TODO
#goa_rgd TODO
#goa_mgi TODO
#goa_xenbase TODO
ortho_mapping_zfin_human data_files/zfin_human_ortholog_mapping.txt https://zfin.org/downloads/human_orthos.txt danio_rerio
ortho_mapping_mgi_human data_files/mgi_human_ortholog_mapping.txt https://www.informatics.jax.org/downloads/reports/HOM_MouseHumanSequence.rpt mus_musculus
ortho_mapping_rgd_human data_files/rgd_human_ortholog_mapping.txt https://download.rgd.mcw.edu/data_release/HUMAN/ORTHOLOGS_HUMAN.txt rattus_norvegicus
ortho_mapping_xenbase_human data_files/xenbase_human_ortholog_mapping.txt https://download.xenbase.org/xenbase/GenePageReports/XenbaseGeneHumanOrthologMapping.txt xenopus
###states_of_interest [SOI name] [to be expressed + or suppressed -]
chronic_inflammation +
cancer +
###categories [category] [True / False]
biological_process True
molecular_activity True
cellular_component False
###GO_terms [GO id] [process] [upregulated + or downregulated - or general 0] [weight 0-1] [GO term name - optional] [GO term description - optional]
GO:0006954 chronic_inflammation + 1 inflammatory response
GO:1900408 chronic_inflammation - 1 negative regulation of cellular response to oxidative stress
GO:1900409 chronic_inflammation + 1 positive regulation of cellular response to oxidative stress
GO:2000524 chronic_inflammation - 1 negative regulation of T cell costimulation
GO:2000525 chronic_inflammation + 1 positive regulation of T cell costimulation
GO:0002578 chronic_inflammation - 1 negative regulation of antigen processing and presentation
GO:0002579 chronic_inflammation + 1 positive regulation of antigen processing and presentation
GO:1900017 chronic_inflammation + 1 positive regulation of cytokine production involved in inflammatory response
GO:1900016 chronic_inflammation - 1 negative regulation of cytokine production involved in inflammatory response
GO:0001819 chronic_inflammation + 1 positive regulation of cytokine production
GO:0001818 chronic_inflammation - 1 negative regulation of cytokine production
GO:0050777 chronic_inflammation - 1 negative regulation of immune response
GO:0050778 chronic_inflammation + 1 positive regulation of immune response
GO:0002623 chronic_inflammation - 1 negative regulation of B cell antigen processing and presentation
GO:0002624 chronic_inflammation + 1 positive regulation of B cell antigen processing and presentation
GO:0002626 chronic_inflammation - 1 negative regulation of T cell antigen processing and presentation
GO:0002627 chronic_inflammation + 1 positive regulation of T cell antigen processing and presentation
GO:0007162 cancer + 1 negative regulation of cell adhesion
GO:0045785 cancer - 1 positive regulation of cell adhesion
GO:0010648 cancer + 1 negative regulation of cell communication
GO:0010647 cancer - 1 positive regulation of cell communication
GO:0045786 cancer - 1 negative regulation of cell cycle
GO:0045787 cancer + 1 positive regulation of cell cycle
GO:0051782 cancer - 1 negative regulation of cell division
GO:0051781 cancer + 1 positive regulation of cell division
GO:0030308 cancer - 1 negative regulation of cell growth
GO:0030307 cancer + 1 positive regulation of cell growth
#GO:0043065 cancer - 1 positive regulation of apoptotic process
#GO:0043066 cancer + 1 negative regulation of apoptotic process
GO:0008285 cancer - 1 negative regulation of cell population proliferation
GO:0008284 cancer + 1 positive regulation of cell population proliferation
The main role of the researcher is to establish one or more custom states of interest (SOIs) and then attribute specific GO terms to the SOIs. Thus, SOIs and GO term attributions will be covered first.
Creating SOIs (states_of_interest section)
States of interest are created in the states_of_interest section. A SOI represents a name of a specific state of interest. Besides the name, either + or - is added in the line beside the SOI name in order to specify whether the researcher is interested in finding genes responsible for the positive contribution (stimulation) of the SOI or the negative contribution (inhibition) of the SOI.
For example, when a researcher observes increased capillary growth in a histological sample, an SOI could be angiogenesis +. Strictly speaking, an SOI is only angiogenesis, whereas the + or - represents the stimulation or inhibition of the SOI. When both the SOI and the direction of regulation of that SOI are specified in the states_of_interest, this is termed a target SOI.
Attributing GO terms to SOIs (GO_terms section)
After SOIs have been created, they need to be attributed with GO terms to specifically define them. SOIs can have GO terms attributed both for stimulation (+) or inhibition (-) of the SOI, irrespective of the defined target SOIs in the states_of_interest section. GO terms are attributed to SOIs in the GO_terms section, by first specifying a GO term id, followed by the SOI, the impact of the GO term on the SOI (+ or -), a weight (this is historical and is kept at 1) and a description of the GO term.
Example: A researcher defined an angiogenesis SOI. Now, the researcher can assign GO terms that positively and negatively stimulate angiogenesis such as:
GO:0016525 angio - 1 negative regulation of angiogenesis
GO:0045766 angio + 1 positive regulation of angiogenesis
GO:0043534 angio + 1 blood vessel endothelial cell migration
GO:0043532 angio - 1 angiostatin binding
With a defined SOI(s) and attributed GO terms, you can actually run the analysis and leave the other options at defaults. Other sections are explained in the following text.
Evidence code groups section
Evidence codes are three- or two-letter codes providing a specific level of proof for an annotation between a GO term and a specific gene. This section contains the whole hierarchy of possible evidence codes, grouped into several major evidence code groups (EGCs). This section only determines the possible EGCs and specific evidence codes, whereas the EGCs or specific evidence codes are selected in the Settings section via the evidence_codes setting.
Based on https://geneontology.org/docs/guide-go-evidence-codes/, there are the following 6 EGCs (noted with belonging evidence codes):
- experimental evidence (EXP, IDA, IPI, IMP, IGI, IEP, HTP, HDA, HMP, HGI, HEP)
- phylogenetically inferred evidence (IBA, IBD, IKR, IRD)
- computational analysis evidence (ISS, ISO, ISA, ISM, IGC, RCA)
- author statement evidence (TAS, NAS)
- curator statement evidence (IC, ND)
- electronic annotation (IEA)
Of important notice is that approximately 95% of Gene Ontology annotations are electronically inferred (IEA) and these are not checked by a human examiner.
This section exists to give user the option to add or exclude any evidence codes, should the GO evidence codes change in the future. Each line contains two tab-separated elements:
- evidence code group name (e.g. author_statement)
- evidence codes (e.g. TAS,NAS) belonging to the group, along with their ECO identifiers (evidence code and identifier separated by underscore) as comma-separated values (e.g. TAS_ECO:0000304,NAS_ECO:0000303)
ECO evidence code identifiers can be found on https://wiki.geneontology.org/index.php/Guide_to_GO_Evidence_Codes and https://www.ebi.ac.uk/QuickGO/term/ECO:0000245.
WARNING: The evidence codes section MUST be specified before the settings section.
Example:
###evidence_code_groups
experimental EXP_ECO:0000269,IDA_ECO:0000314,IPI_ECO:0000353,IMP_ECO:0000315,IGI_ECO:0000316,IEP_ECO:0000270,HTP_ECO:0006056,HDA_ECO:0007005,HMP_ECO:0007001,HGI_ECO:0007003,HEP_ECO:0007007
phylogenetic IBA_ECO:0000318,IBD_ECO:0000319,IKR_ECO:0000320,IRD_ECO:0000321
computational_analysis ISS_ECO:0000250,ISO_ECO:0000266,ISA_ECO:0000247,ISM_ECO:0000255,IGC_ECO:0000317,RCA_ECO:0000245
author_statement TAS_ECO:0000304,NAS_ECO:0000303
curator_statement IC_ECO:0000305,ND_ECO:0000307
electronic IEA_ECO:0000501
Settings section
The settings section contains several settings, which are used to change the flow of the algorithm.
evidence_codes is used to determine which annotations between GO terms and respective genes the algorithm will accept. GOReverseLookup will only accept genes annotated to input GO terms with any of the user-accepted evidence codes.
- to accept all evidence codes belonging to a specific EGC, use a tilde operator in brackets
(~), e.g.experimental(~) - to accept specific evidence codes belonging to an evidence group, specify them between the parentheses. If specific evidence codes are specified among parantheses, all non-specified evidence codes will be excluded. For example, to take into account only IC, but not ND, from curator_statement, use the following:
curator_statement(IC) - to exclude specific evidence codes, use an exclamation mark. All evidence not specified excluded evidence codes belonging to an EGC will still be included. To exclude only HEP and retain the rest of experimental evidence codes, use:
!experimental(HEP) - to merge multiple evidence code groups, supply them as comma-separated values. E.g.:
experimental(~),phylogenetic(~),computational_analysis(~),author_statement(TAS),curator_statement(IC),!electronic(~)
Example evidence codes:
evidence_codes experimental(~),phylogenetic(~),computational_analysis(~),author_statement(TAS),!curator_statement(ND),!electronic(~)
pvalue is the threshold p-value used to assess the statistical significance of a gene being involved in a target SOI. There are two possible cases of evaluation:
a) The user has defined an SOI and has attributed GO terms that both positively and negatively regulate the SOI. A gene is statistically significant if its p-value for the defined SOI stimulation/inhibition is less than the defined p-value threshold AND its p-value for the opposite SOI (inhibition/stimulation) is greater than the defined p-value threshold. It is advisable to also attribute GO terms that are opposite regulators of the defined target SOI in order to increase the credibility of the results.
b) The user has defined an SOI and has attributed GO terms only in one regulation direction (e.g. only stimulation or only inhibition). A gene is statistically significant if its p-value for the defined SOI is less than the defined p-value threshold.
target_organism is the target organism for which the statistical analysis is being performed. Organisms are represented with three identifiers (separated by vertical bars), which MUST be supplied for the program to correctly parse organism data: (1) organism label in lowercase (2) organism database and (3) organism NCBI taxon. For example, to select Homo sapiens as the target organism, a researcher would specify:
target_organism homo_sapiens|UniProtKB|NCBITaxon:9606
multiple_correction_method: the multiple correction method used in statistical evaluation of significant genes. Default is fdr_bh. Must be one of the following:
bonferroni: one-step correctionsidak: one-step correctionholm-sidak: step down method using Sidak adjustmentsholm: step-down method using Bonferroni adjustmentssimes-hochberg: step-up method (independent)hommel: closed method based on Simes tests (non-negative)fdr_bh: Benjamini/Hochberg (non-negative)fdr_by: Benjamini/Yekutieli (negative)fdr_tsbh: two stage fdr correction (non-negative)fdr_tsbky: two stage fdr correction (non-negative)
Also refer to: https://www.statsmodels.org/dev/generated/statsmodels.stats.multitest.multipletests.html#statsmodels.stats.multitest.multipletests
two_tailed: If True, it will find significant genes at both tails of the significance curve. The chosen significant genes depend on amount of SOIs defined by the user, namely:
- if only a target SOI is defined by the researcher in the input file, without its corresponding reverse SOI (e.g.
tumorigenesis+, denoting stimulation of tumorigenesis, withouttumorigenesis-, denoting inhibition of tumorigenesis). Supposepis set to 0.05. In this case, the program finds genes significant in the stimulation of tumorigenesis, which satisfy the conditionp(tumorigenesis+) < 0.05. The program also artifically creates a reverse SOI (in this case,tumorigenesis-), finds genes which satisfyp(tumorigenesis+) > 0.95, and associates such genes with the reverse SOI. The output file contains both the defined target SOI (tumorigenesis+) and its (albeit artifically created by the program) reverse SOI (tumorigenesis-), along with significant genes for each SOI. - if a researcher defined target SOIs and complementary reverse SOIs in the input file, a gene is significant, if it is significantly associated with all target SOIs (p < 0.05) and insignificantly associated with all reverse SOIs (p >= 0.05). If two-tailed setting is True, then the program also evaluates genes, which are significantly associated with all reverse SOIs (p < 0.05) and insignificantly associated with all target SOIs (p >= 0.05).
ortholog_organisms represent all homologous organisms, the genes of which are also taken into account during the scoring phase if they are found to have existing target organism orthologous genes. This feature has been enabled as a GO term can be associated with genes belonging to different organisms, which are indexed by various databases. The current model has been tested on the following orthologous organisms: Rattus norvegicus, Mus musculus, Danio rerio and Xenopus tropicalis. Example:
ortholog_organisms danio_rerio|ZFIN|NCBITaxon:7955,rattus_norvegicus|RGD|NCBITaxon:10116,mus_musculus|MGI|NCBITaxon:10090,xenopus_tropicalis|Xenbase|NCBITaxon:8364
include_indirect_annotations: The first parameter (True or False) determines whether to enable this setting. Enabling indirect annotations means increasing the amount of annotations to a gene by the sum of all indirectly annotated GO terms (obtained by traversing the GO term hierarchy tree using the directly annotated GO terms to the gene in question). The second parameter (p or c) determines whether to obtain indirect annotations as parents (p) or as children (c) of the directly annotated GO terms.
As defined in GO, the "true" indirect annotations between a gene and a directly annotated GO term are the parents of the directly annotated GO term, whereas the children GO terms of the directly annotated GO term are not necessarily regulated by the gene in question. However, when studying the impact of a gene on the regulation of a given SOI, the reverse holds true - the children of the directly annotated GO term are the ones that also regulate an SOI, whereas it cannot be claimed so for the parent GO terms.
Consider the following tree:
GO:2000026 regulation of multicellular organismal development
- GO:1901342 regulation of vasculature development
- GO:0045765 regulation of angiogenesis
- GO:0045766 positive regulation of angiogenesis <- gene Hipk2
- GO:1905555 positive regulation of blood vessel branching
- GO:1903672 positive regulation of sprouting angiogenesis
- GO:0035470 positive regulation of vascular wound healing
- GO:0016525 negative regulation of angiogenesis
Gene Hipk2 is directly annotated to GO:0045766. All the parent GO terms also infer the annotation to the gene Hipk2 (GO:0045765, GO:1901342, GO:2000026), but not the child terms (GO:1905555, ...). However, if the defined target SOI by the researcher is 'stimulated angiogenesis', then the GO terms responsible for the upregulation of angiogenesis are actually the children terms of GO:0045766, rather than the more non-specific parent terms. For example, the term "regulation of angiogenesis" would be faultily counted as stimulatory to angiogenesis during the gene scoring process, as it also encompasses a "negative regulation of angiogenesis" child term (among others).
The predicament in "gene influence studies" is thus whether to use parent or child terms as indirect annotations. Parent terms definitely hold the annotation to a gene of a directly annotated GO term, but are less specific in regulating a given target SOI. Child terms are more specific in regulating a given target SOI (which is desired in gene influence studies), however the connection between the child terms and the directly annotated GO term is made based on the assumption that all child terms regulate the same SOI as the directly annotated term (which the parents term might not, or might be too vaguely defined). Still, the gene in question might not be associated with any of the child terms.
Due to the aforementioned dilemma, a researcher can choose whether to count parents or children as indirect annotations.
indirect_annotations_max_depth takes an integer value as a parameter, with the value of -1 meaning "infinite depth". When querying indirect annotations, specifically parent annotations, the GO terms very high in the hierarchy tree are shared across all genes in a research model (e.g. "biological regulation", "biological process", ...). To prevent such vague terms from faultily influencing the scoring process of genes, a user can set a fixed maximum depth of indirect annotations that are used for the scoring process. Consider the following example:
GO:0008150 biological_process
- GO:0048731 system_development
- GO:0001944 vasculature_development
- GO:1901342 regulation_of_vasculature_development
- GO:0022603 regulation_of_anatomical_structure_morphogenesis
- GO:0045764 regulation_of_angiogenesis
- GO:0045766 positive_regulation_of_angiogenesis
If the user were querying parents as indirect annotations without a maximum depth limit, all indirect annotations up to the root term would have influenced the gene statistical relevance. However, if a user set the maximum depth limit to 3 (via indirect_annotations_max_depth 3), then only the closest three indirect annotations would have been considered (e.g. GO:0045764, GO:0022603 and GO:1901342).
goterm_gene_query_timeout is the timeout it takes when querying genes annotated to GO terms. If specifying very vague GO terms (such as regulation of gene expression, which has ~25 million annotations, a query might fail due to a request taking too long to complete or, which is a more severe error due to its covertness, a query might return an incomplete list of genes associated with a GO term. As a rule of thumb, we discourage the usage of such vague GO terms. A default 240-second timeout ensures that all GO terms approximately with a few million annotations are fetched correctly from the GO servers.
goterm_gene_query_max_retries is the maximum number of retries sent to the GO servers before dropping a GO term and assigning it with an empty list of associated genes.
gorth_ortholog_refetch We implemented a gOrth batch ortholog query (https://biit.cs.ut.ee/gprofiler/orth), which speeds up the total runtime of the program. The function attempts to find orthologs to genes in a single batch request. If 'gorth_ortholog_refetch' is True, then the genes for which orthologs were not found will be re-fetched using alternative Ensembl calls. If 'gorth_ortholog_refetch' is False, then the genes for which orthologs were not found will not be queried for orthologs again.
gorth_ortholog_fetch_for_indefinitive_orthologs The gOrth batch query implementation can return the following options:
- multiple orthologous genes (these are called "indefinitive orthologs")
- a single orthologous gene (called a "definitive ortholog")
- no orthologous genes.
In our asynchronous Ensembl ortholog query pipeline implementation, when multiple orthologous genes are returned from Ensembl, the orthologous gene with the highest percentage identity (percentage identity of amino-acid sequence between the gene and the target organism orthologous gene) is selected as the best ortholog and is assigned as the true ortholog to the input gene. However, gOrth has currently (10_29_2023) no option to return the "best" orthologous gene, neither it has the option to exclude obsolete ortholog gene ids (confirmed by the gProfiler team via an email conversation). Therefore, it is advisable to keep the gorth_ortholog_fetch_for_indefinitive_orthologs to True, so that indefinitive orthologs are discarded from the gOrth ortholog query and are instead fetched by the asynchronos pipeline, which can select the best ortholog for the input gene. Having this setting set to False will choose, in the case of indefinitive orthologs, the first returned ortholog id from the gOrth query, but with no guarantees that this ortholog id is not obsolete.
fisher_test_use_online_query It is highly advisable to leave this setting set to False, otherwise, the timing of the scoring phase might severely be extended (into days, if not weeks).
uniprotkb_genename_online_query: When querying all genes associated to a GO Term, Gene Ontology returns UniProtKB identified genes (amongst others, such as ZFIN, Xenbase, MGI, RGD). During the algorithm, gene name has to be determined. It can be obtained via two pathways:
- online pathway, using UniProtAPI
- offline pathway, using the GO Annotations File
During testing, it has been observed that the offline pathway usually results in more gene names found, besides being much faster. Thus, it is advisable to leave this setting set to False, both to increase speed and accuracy. If it is set to True, then gene names will be queried from the UniProtKB servers.
Asynchronous settings Several settings influence the speed of asynchronous web requests being sent to the servers. If the speed exceeds server thresholds, however, the server may start to block incoming requests from your computer. Thus, if you experience blocking, the suggested asynchronous settings parameters are the following:
goterm_name_fetch_async False
goterm_gene_fetch_async False
goterm_name_fetch_req_delay 1.5
goterm_name_fetch_max_connections 5
goterm_gene_fetch_req_delay 0.8
goterm_gene_fetch_max_connections 5
Filepaths section
The filepaths section specifies several files that will be used during the program's runtime. Each file is represented in a single line by four parameters: (1) the file label (e.g. goa_human), (2) relative path to the file (e.g. data_files/goa_human.gaf), (3) the file download url (e.g. http://geneontology.org/gene-associations/goa_human.gaf.gz) and (4) the organism label pertaining to the file (e.g. homo_sapiens). We suggest beginner users NOT to change anything in the filepaths section. An example filepaths section is:
###filepaths
go_obo data_files/go.obo https://purl.obolibrary.org/obo/go.obo all
goa_human data_files/goa_human.gaf http://geneontology.org/gene-associations/goa_human.gaf.gz homo_sapiens
ortho_mapping_zfin_human data_files/zfin_human_ortholog_mapping.txt https://zfin.org/downloads/human_orthos.txt danio_rerio
ortho_mapping_mgi_human data_files/mgi_human_ortholog_mapping.txt https://www.informatics.jax.org/downloads/reports/HOM_MouseHumanSequence.rpt mus_musculus
ortho_mapping_rgd_human data_files/rgd_human_ortholog_mapping.txt https://download.rgd.mcw.edu/data_release/HUMAN/ORTHOLOGS_HUMAN.txt rattus_norvegicus
ortho_mapping_xenbase_human data_files/xenbase_human_ortholog_mapping.txt https://download.xenbase.org/xenbase/GenePageReports/XenbaseGeneHumanOrthologMapping.txt xenopus
A brief explanation of the files:
go.obo is a Gene Ontology file representing the entire GO term hierarchy tree. It is used in the scoring phase of the GOReverseLookup's algorithm in order to obtain indirectly annotated (children) GO terms of directly annotated GO terms to a specific gene.
goa_human.gaf is a Gene Ontology Annotations file and represents the annotations between genes and GO terms for a specific organism. It is used during the scoring phase of the GOReverseLookup's algorithm to obtain the number of all GO terms from the entire Gene Ontology associated with a given gene for a given organism. The GAF file used in the scoring to obtain the aforementioned GO term count should be constructed for the organism, which the research investigates. Currently, only the human GAF can be used and thus GOReverseLookup is currently limited only to research for the Homo sapiens species, but we plan to introduce full GAF modularity, so that the user will be able to supply a GAF file for any desired organism.
3rd party database files are some non-UniProtKB files that are also used for faster orthologous gene queries. Currently supported organisms are Danio rerio, Rattus norvegicus, Xenopus tropicalis and Mus musculus. The user should not change these. The support for these database files does not limit the amount of orthologous organisms a user can add via the ortholog_organisms setting.
ortho_mapping_zfin_human data_files/zfin_human_ortholog_mapping.txt https://zfin.org/downloads/human_orthos.txt danio_rerio
ortho_mapping_mgi_human data_files/mgi_human_ortholog_mapping.txt https://www.informatics.jax.org/downloads/reports/HOM_MouseHumanSequence.rpt mus_musculus
ortho_mapping_rgd_human data_files/rgd_human_ortholog_mapping.txt https://download.rgd.mcw.edu/data_release/HUMAN/ORTHOLOGS_HUMAN.txt rattus_norvegicus
ortho_mapping_xenbase_human data_files/xenbase_human_ortholog_mapping.txt https://download.xenbase.org/xenbase/GenePageReports/XenbaseGeneHumanOrthologMapping.txt xenopus
Categories section
Gene Ontology provides three categories of annotations (as known as Gene Ontology Aspects):
- molecular_activity
- biological_process
- cellular_component
The categories section allows you to determine which GO Terms will be queried either from online or from the GO Annotations File. For example, when a researcher is only interested in GO Terms related to molecular activity and biological processes, querying GO Terms related to a cellular component might result in an incorrect gene scoring process, resulting in some genes being scored as statistically insignificant, whereas they should be statistically significant. Thus, a researcher should turn off or on the GO categories according to the research goals. To turn on or off a specific GO category, provide a tab-delimited True or False value next to that category. Example:
###categories [category] [True / False]
biological_process True
molecular_activity True
cellular_component False
Running the program
Once the input file is complete, it is time to run the program using the following steps:
- activate the Python's virtual environment (as instructed in Creating your GOReverseLookup workspace). To recap: (1) open the command-prompt (2) pass the filepath to the
.../goreverselookup/Scripts/activateto activate your virtual environment. By activating the virtual environment, the base working directory for the program will be set to.../goreverselookup/. A curious reader might have observed that in the input file, data file paths are specified in relative notation (e.g.data_files/go.obo) - they are relative to the base working directory. By activating the virtual environment, you ensure both that the GOReverseLookup is correctly installed and that all files in use or created by the GOReverseLookup program are saved to the.../goreverselookup/folder. The result of activation should look something like this:
-
switch the command prompt's current working directory (CWD) to the CWD of GOReverseLookup's virtual environment: If using Windows, use the cd (change directory) command to navigate to the root folder of the Python's virtual environment where GOReverseLookup is installed:
cd .../goreverselookup(e.g.cd C:\Users\User\Development\goreverselookup). Setting the CWD is important, as the CWD set in the command prompt will also be the current working directory of the GOReverseLookup program. Thus, if no CWD is set and the user supplies an input file to the program (e.g.research_models/input.txt), the program cannot find the input file, as all relative filepath queries are performed from the CWD. Setting the CWD also ensures that the final analysis results of a GOReverseLookup analysis will be saved to a correct location. -
run GOReverseLookup with either of the commands:
goreverselookup PATH_TO_INPUT_FILEorgoreverselookup PATH_TO_INPUT_FILE PATH_TO_OUTPUT_FOLDER(e.g.goreverselookup "research_models/input.txt"orgoreverselookup "research_models/input.txt" "results"). When supplying thePATH_TO_OUTPUT_FOLDERparameter, also create the output folder inside the.../goreverselookup/folder. When only the input file is specified, analysis results will be saved into the same base folder where the input file resides. Thus, if the input file resides in...goreverselookup/research_models/input.txt, results will be saved to.../goreverselookup/research_models/folder.
If you have created multiple input.txt files in your directory structure (e.g. you want to analyze multiple research models), then use the full directory operation parameter, to which you pass the absolute path to the root directory, where the program should start searching for all the input.txt files:
goreverselookup "input.txt" --full_directory_op "C:/.../goreverselookup/"
If different research models share the same GO terms to SOI mappings and the same evidence codes, and the difference between them is only in the desired p-values or the inclusion/exclusion of indirect annotations, then you can use --rescore True to only use the first analyzed research model as the base model, which is scored against different scoring criteria (p-value, indirect annotations) of other research models under the same root directory. This significantly reduces runtime, since it avoids research model reanalysing. Example:
goreverselookup "input.txt" --full_directory_op "C:/.../goreverselookup/" --rescore True
- wait for GOReverseLookup to complete the analysis
WARNING: When the scoring phase of the program is completed, 3-5 minutes will elapse for the saving of the cache files to complete. Do not close the command-prompt during this time, otherwise the cache files will be corrupt. Cache files are useful during recurrent runs of the program, as they prevent re-querying for the results of the same GO Terms or genes that have already been queried.
WARNING: A sign of cache file corruptness are usually JSON errors that occur during the beginning of a GOReverseLookup anaylsis. You can fix this by manually deleting the cache folder located at .../goreverselookup/cache/.
When using asynchronous querying for GO term products, if one of the requests inside a batch of requests exceeds the 'goterm_gene_query' timeout value (one of the settings), the entire batch of product queries will fail. This usually happens when the user attempts to collect products of GO terms with millions of more annotated genes. For us, an experimental 'goterm_gene_query' timeout value that successfully queries GO terms with ~1 million annotated genes is 240 seconds.
Analysing the program results in a text editor
When GOReverseLookup analysis is finished, two distinct JSON files will be saved:
data.json: This file represents the entire knowledge about the constructed research model, with all statistically significant and insignificant genesstatistically_relevant_genes.json: This file represents the discovered statistically significant genes.
We suggest downloading a rich text editor, such as Notepad++, which uses syntax highlighting to make the JSON files more readable and also allows the user to collapse sections of the JSON file. Example result - a statistically significant gene named IL6 was found to be statistically relevant in stimulating chronic inflammation and cancerous cell growth:
{
"chronic_inflammation+:cancer_growth+": [
{
"id_synonyms": [
"MGI:96559",
"ENSMUSG00000025746",
"ENSRNOG00000010278",
"UniProtKB:A0A803JUX3",
"ENSXETG00000049395",
"RGD:2901",
"UniProtKB:P05231",
"Xenbase:XB-GENE-480186"
],
"taxon": "NCBITaxon:10090",
"target_taxon": null,
"genename": "IL6",
"description": "interleukin 6",
"uniprot_id": "UniProtKB:P05231",
"ensg_id": "ENSG00000136244",
"enst_id": "ENST00000258743",
"refseq_nt_id": null,
"mRNA": null,
"scores": {
"fisher_test": {
"chronic_inflammation+": {
"n_prod_SOI": 13,
"n_all_SOI": 95,
"n_prod_general": 90,
"n_all_general": 30592,
"expected": 0.2794848326359832,
"fold_enrichment": 46.51415204678363,
"pvalue": 1.4374380950725201e-18,
"odds_ratio": 62.63224580297751,
"pvalue_corr": 1.1302000766317196e-14
},
"chronic_inflammation-": {
"n_prod_SOI": 1,
"n_all_SOI": 62,
"n_prod_general": 90,
"n_all_general": 30592,
"expected": 0.18240062761506276,
"fold_enrichment": 5.482437275985663,
"pvalue": 0.16710866475397615,
"odds_ratio": 5.607109965002763,
"pvalue_corr": 1.0
},
"cancer+": {
"n_prod_SOI": 7,
"n_all_SOI": 37,
"n_prod_general": 90,
"n_all_general": 30592,
"expected": 0.10885198744769874,
"fold_enrichment": 64.30750750750751,
"pvalue": 1.4406227714406763e-11,
"odds_ratio": 85.66425702811244,
"pvalue_corr": 1.1104941767381825e-08
},
"cancer-": {
"n_prod_SOI": 2,
"n_all_SOI": 25,
"n_prod_general": 90,
"n_all_general": 30592,
"expected": 0.07354864016736401,
"fold_enrichment": 27.19288888888889,
"pvalue": 0.0024570992466771188,
"odds_ratio": 30.117588932806324,
"pvalue_corr": 0.05485289192766472
}
}
}
}
]
}
Generating an Excel file from the program results
Suppose the aforementioned file structure:
- goreverselookup/
- research_models/
- test_model/
- input.txt
After analysis is ran by activating the goreverselookup virtual environment and running the command goreverselookup "research_models/test_model/input.txt", a results folder will be saved into test_model, containing two files:
data.json: represents the entire research modelstatistically_relevant_genes.json: represents the discovered statistically significant genes (which align with the defined target SOIs specified by the research model)
Therefore:
- goreverselookup/
- research_models/
- test_model/
- input.txt
- results/
- data.json
- statistically_relevant_genes.json
To view the discovered statistically relevant genes, run the command goreverselookup PATH_TO_STAT_RELEVANT_GENES --report True, e.g. goreverselookup research_models/test_model/results/statistically_relevant_genes.json --report True. This will first print out a report of tab-separated-values to the console, such as:
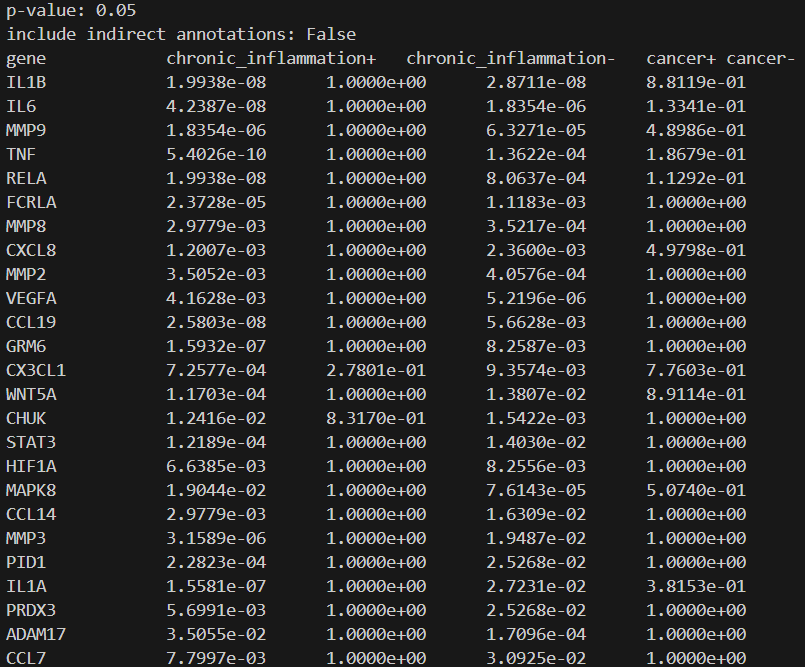
If you want to analyze all statistically relevant genes files in a given directory (and all subfolders), then use the full directory operation command, after which you specify the root directory (the absolute path to the starting directory where the program should start traversing the file structure and thus searching for the statistically relevant genes files):
goreverselookup "statistically_relevant_genes.json" --report True --full_directory_op <ROOT_DIR>
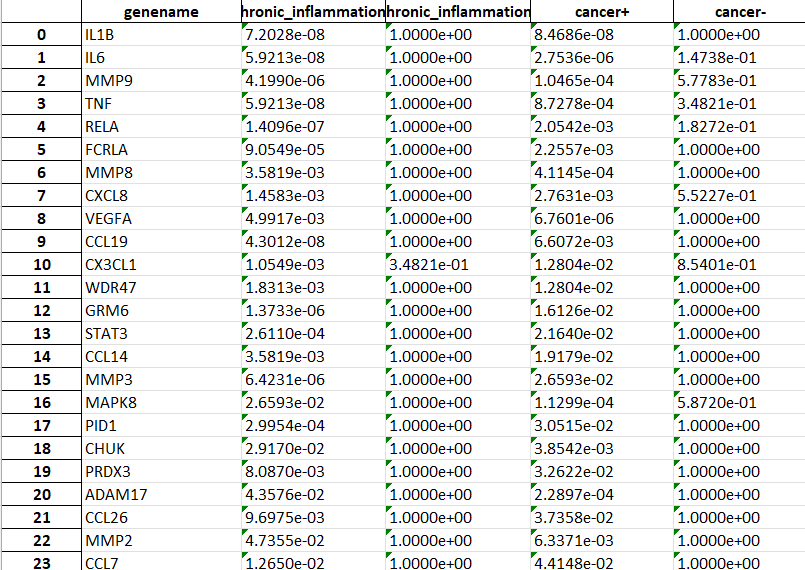
Additionally, an Excel file will be generated inside results:
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file goreverselookup-1.0.77.tar.gz.
File metadata
- Download URL: goreverselookup-1.0.77.tar.gz
- Upload date:
- Size: 169.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1c3259558f25c71b2b9bc8e4a0e7206649d251085c2df646e896c35aec87aefa
|
|
| MD5 |
485fb03ab0dcf279aa3eeede967de905
|
|
| BLAKE2b-256 |
ae4faeed3387c07ce03e4930d73d128ee1e58f62a835a8444d2cdb7a74c39889
|
File details
Details for the file goreverselookup-1.0.77-py3-none-any.whl.
File metadata
- Download URL: goreverselookup-1.0.77-py3-none-any.whl
- Upload date:
- Size: 175.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d223e5c2162b0d8f4e465974833d43488f859a5d12581937957cdf4a046dbd9d
|
|
| MD5 |
22a2496bdcd5defb1ea8bc44c1f6738b
|
|
| BLAKE2b-256 |
0a5e69f4d388f9e47a464140ba602eeb0d8f054f0cf8fce2bb78947dac835865
|







