A python package designed to communicate among various chemical and materials calculational tools

Project description

🥘Hotpot(火锅): AI-Driven Infrastructure for Chemistry
Bridging the gap between Chemical Intuition and Artificial Intelligence. From Empirical Rules to Data-Driven Foundation Models.
In Hotpot, every ingredient is cookable. 什么都能涮
In Data-Driven, every problem is computable. 什么都能算
(The Chinese phrases are just the Chinese versions of the English lines. “涮 / shuàn” (“to dip in hotpot”) and “算 / suàn” (“to compute”) form a wordplay because of their similar sound.)
Contents
📖 Introduction
Hotpot is not just a chemical informatics toolkit; it is a research-grade infrastructure designed to digitize, model, and analyze chemistry environments.
Unlike traditional tools (e.g., RDKit, OpenBabel) that rely heavily on explicit valence rules—which often fail in metal-ligand scenarios—Hotpot adopts a Data-Driven Philosophy. It seamlessly integrates a robust chemical kernel with modern deep learning pipelines, enabling "Fuzzy Modeling" for complex chemical intuition that cannot be captured by simple mathematical formulas.
Crucially, Hotpot abstracts the complexity of Artificial Intelligence into a silent, high-performance backend.
To the user, Hotpot feels like the familiar tools you already use. It simulates manipulating actual
chemical entities -- whether a single Molecule or a periodic Crystal Lattice. You interact solely with
intuitive Molecule and Crystal objects—the standard vernacular of chemistry. The massive AI training
frameworks and complex inference engines run entirely behind the scenes, invisible and automated.
- Minimal-AI Code: Users typically do not need to touch tensors, write training loops, or configure neural networks.
- Seamless Adaptation: For standard tasks, the default models work out of the box. For specific domains, you simply organize your private data into Molecule objects; Hotpot ingests the data and refines the engine automatically.
🏗️ Key Features & Architecture
Hotpot is built on a modular architecture designed to hide complexity. It consists of a robust Chemical Kernel for data handling and a silent AI Engine for intelligence.
1. The Chemical Kernel (hotpot.core)
The robust foundation that digitizes chemistry.
- Chemist-Centric Interface:
- Intuitive Operations: Operates in the natural vernacular of chemistry. You interact with
Molecule,Atom, andBondobjects directly—manipulating structures in code feels exactly like building models in a lab. - Plays nicely with existing cheminformatics tools and workflows, preserving the interfaces users are already used to.
- Intuitive Operations: Operates in the natural vernacular of chemistry. You interact with
- Multi-Scale Property Integration:
- Micro to Macro: A unified interface for managing diverse physical properties. Effortlessly manage microscopic descriptors
(
Atom.elements,Molecule.descriptors) alongside macroscopic observables (Molecule.get_thermo()).
- Micro to Macro: A unified interface for managing diverse physical properties. Effortlessly manage microscopic descriptors
(
- Universal I/O Bridge:
- Read/Write Common Formats: Seamlessly handles standard chemistry formats such as
.mol2,.cif,.xyz, and Gaussian.gjf. - AI-Ready Graphs: Transparently converts structures into graph representations suitable for modern deep learning models, without exposing low-level details to the user.
- Read/Write Common Formats: Seamlessly handles standard chemistry formats such as
2. Data-Driven Analysis (Pre-trained & Ready)
Intelligence baked into the Molecule object (especially, for Coordination Chemistry).
- Coordination pattern determination:
AIModel.cbond: Surpasses traditional valence rules by using deep learning to predict coordinate bonds in complex transition metal environments.
- 3D Structure Initialization (
complexes_build_optimize_):- AI-refined 3D build: A specialized pipeline for generating metal complexes with AI assisting
- Topology-aware optimization: Adds continuous topological inspection during geometry optimization and applies tailored breaking / reconstruction strategies, preventing common failures in metal complex 3D generation, such as tangled chain, interlocked rings, and other non-optimizable artifacts.
- Connecting microscopic models with macroscopic observables:
- Macroscopic properties (
logβ,logD, ...) are typically statistical constructs emerging from ensembles of microscopic states, rather than from any single configuration. Relying on a small number of static microscopic models to infer macroscopic behavior can therefore introduce substantial bias and be misleading. - Hotpot combines approximate microscopic models with rich molecular representations and environmental variables to make this micro–macro connection more reliable. Embracing the idea that “all models are wrong, but some are useful”, Hotpot uses AI-based fuzzy modeling to improve the robustness and accuracy of inferring macroscopic observables from microscopic model.
- Macroscopic properties (
- Oxidation state identification - coming soon ...
- Other important chemical problems
- If there is a core chemistry task you think should be “built-in” to the
Moleculeobject, feel free to open an issue and describe your use case.
- If there is a core chemistry task you think should be “built-in” to the
3. Assembly & Generation of Virtual Molecules
From fragment-based enumeration to AI-driven molecular design.
-
High-throughput fragment-based assembly
- Assemble virtual molecules from scaffolds and fragments at scale, enabling grid-like exploration of targeted chemical spaces (e.g. focused libraries around a given scaffold or motif).
-
AI-based molecular generation
- Molecular generation: Generate new candidate molecules by learning from a small set of example structures, proposing novel analogues in the same “chemical family” or design space.
- Conditional molecular generation: Generate molecules under explicit goals or constraints — e.g. guided by target properties, property predictors, or user-defined objective functions — to search for structures that optimize (maximize / minimize) desired performance while respecting structural patterns of the examples.
4. Optimization of Wet Experiments
Close the loop between computation and lab experiments.
-
Multiple optimization strategies
- Supports a range of optimization backends, including Bayesian optimization (BO) and evolutionary algorithms (EA), for efficient exploration of experimental parameter spaces.
-
Structure-aware experimental optimization
- Combines experimental parameters with optional structural / molecular representations, enabling joint optimization over both reaction conditions and molecular features.
-
Mixed-type design spaces
- Handles continuous and discrete variables in a unified framework, suitable for real experimental design problems (temperatures, pH, solvents, ligands, catalysts, etc.).
-
Manifold / parameter-space visualization
- Provides visualization of the explored parameter manifold and optimization trajectory to help chemists understand where the optimizer is searching and why.
-
CLI integration
- Exposed via a simple command-line interface, e.g.
hotpot optimize ..., so optimization workflows can be scripted and automated without additional boilerplate.
- Exposed via a simple command-line interface, e.g.
📥 Installation
Requirements
- python == 3.9 *
- openbabel >= 3.1.1
- cclib
- lammps
- onnxruntime
* Note: Hotpot strictly requires Python 3.9 due to specific regex behaviors and C++ binding compatibility in the underlying chemical kernel. Upgrading to 3.10+ may cause parsing errors in legacy molecular formats.
1. Install dependencies
Before installing Hotpot, you should install its dependencies first. It is
recommended to create a new conda environment to run the package.
conda create -n hp python==3.9 openbabel cclib lammps onnxruntime -c conda-forge
conda activate hp
2. Install
PyPI (Recommended)
pip install hotpot-zzy
Source
git clone https://github.com/Zhang-Zhiyuan-zzy/hotpot.git
pip install build # install `build` package
python -m build
pip install dist/hotpot_zzy-`VERSION`-py3-none-any.whl
📌 Usage examples
1.Building a metal-ligand pair
import hotpot as hp
smi = 'O=C(N(C)CCC)C(C=C1)=NC2=C1C=CC3=C2N=C(C4=NC(C(C)(C)CCC5(C)C)=C5N=N4)C=C3' # (CyMe4)Pyz-PrMe-DIPhen extractant
ligand = hp.read_mol(smi)
pair = ligand.auto_pair_metal('Eu')
print(pair.smiles)
Generate 3D coordinates using complexes_build_optimize_ method
print(pair.coordinates)
pair.complexes_build_optimize_()
print(pair.coordinates)
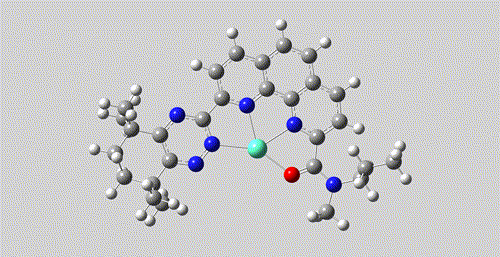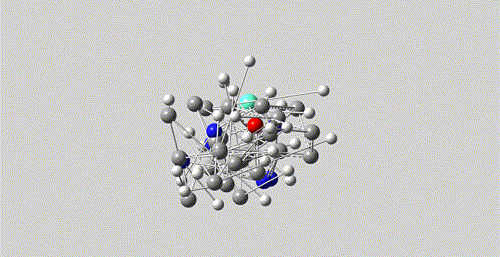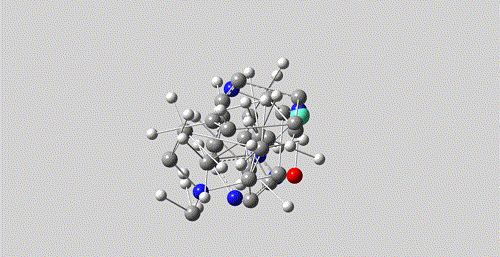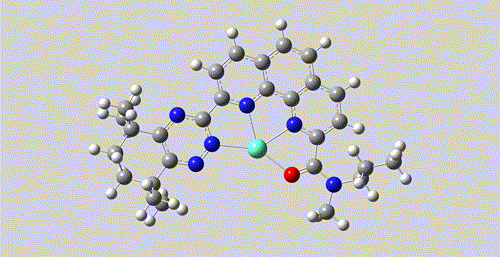
pair.write('./Eu-pair.mol2')
The mol2 file and movie after coordination generation.
Both the formation of coordination bond and the generation of 3D structure are driven by AI model, rather than heuristic rules or pure force fields.
2.Cheminformatics support
The Molecule object is designed to be a familiar, standard cheminformatics tool for chemists.
You can access the Atom, Bond, Rings, and fragment Molecule objects directly through
the properties of Molecule.
Properties
Continuing with the Eu-ligand pair example:
print(pair.atoms)
print(pair.bonds)
print(pair.rings) # all rings
print(pair.ligand_rings) # rings in ligand
assert len(pair.components) == 1
pair.hide_metal_ligand_bonds() # Hide the coordination bonds temporarily
assert len(pair.components) == 2 # Now appears as two fragments: [ligand, metal]
pair.recover_hided_metal_ligand_bonds()
assert len(pair.components) == 1 # Restored to a whole pair
eu_metal = pair.metals[0]
print(eu_metal.neighbours) # [Atom(N), Atom(N), Atom(N), Atom(O)]
print(pair.link_matrix) # Connectivity graph table
SMARTS Support & Extensions
Searching for coordination centers using SMARTS patterns:
hits = pair.search_substructure('[Ln](n)(n)(n)O') # [Ln] --> lanthanide
print(len(hits)) # == 1
print(hits[0].atoms) # [Atom(N32), Atom(O0), Atom(Eu67), Atom(N10), Atom(N17)]
hits = pair.search_substructure('[Ln](n)(n)O')
print(len(hits)) # == 3
hits = pair.search_substructure('[An](n)(n)(n)O') # [An] --> actinide
print(len(hits)) # == 0
Hotpot features a built-in SMARTS parser (API) designed for efficient substructure matching. It supports a subset of the standard SMARTS syntax, covering the majority of atom/bond primitives and first-level logical operators (AND/OR).
Note: Complex nested logic and recursive environments (e.g., recursive SMARTS
$(...)) are not currently supported.
To specifically address the demand in Coordination Chemistry, the syntax has been extended with custom wildcards for metals and periodic table properties:
| Symbol | Definition | Description | Example |
|---|---|---|---|
M |
Metal | Matches any metal atom | [M]~[O] |
!M |
Non-Metal | Matches any non-metal atom | [!M] |
Ln |
Lanthanide | Matches Lanthanide series (La-Lu) | [Ln](n)(n)(n) |
An |
Actinide | Matches Actinide series (Ac-Lr) | [An]~[O] |
NP<n> |
Period | Matches elements in Period n (supports ranges) | [NP4], [NP3-5] |
NG<n> |
Group | Matches elements in Group n (supports ranges) | [NG1], [NG1-2] |
Conversion with RdKit and OpenBabel
Interfacing with other cheminformatics tools:
obMol = pair.to_obmol()
rdMol = pair.to_rdmol()
Converting to PyG (PyTorch Geometric) Data:
data = pair.to_pyg_data()
print(data.x) # Tensor of atom attributes
print(data.x_names) # atom attribute name
print(data.edge_index)
print(data.edge_attr)
print(data.edge_attr_names)
print(data.pair_index) # atom pairs indices
print(data.pair_attr) # pair attrs
print(data.pair_attr_names)
print(data.rings_node_index)
print(data.rings_attr) # Tensor with shape [rings_num, 2]
print(data.rings_attr_names) # ['is_aromatic', 'has_metal']
print(data.rings_node_nums) # How many atoms in a ring
print(data.mol_rings_node_nums) # How many rings in the molecule
print(data.coordinates)
See the cheminfo.core API Documentation for more details.
3.Molecular properties, descriptors, and representation
Extracting thermodynamic properties using thermo library:
import hotpot as hp
mol = hp.read_mol('c1ccc(O)cc1', 'smi') # read a phenol by SMILES
thermo = mol.get_thermo(temp=298.15, pressure=101325)
print(thermo.Tc) # the critical temperature (K)
print(thermo.Psat) # the saturation vapor pressure
print(...)
Extracting the Graph-Spectral representation:
import hotpot as hp
mol1 = hp.read_mol('c1ccc(O)cc1', 'smi')
mol2 = hp.read_mol('c1ccccc1C(=O)O', 'smi')
mol1_ = hp.read_mol('c1ccccc1O', 'smi') # Same molecule, different atom ordering
spectral1 = mol1.graph_spectral()
spectral2 = mol2.graph_spectral()
spectral1_ = mol1_.graph_spectral()
similarity_diff = spectral1 | spectral2
print(similarity_diff) # Similarity in graph spectrum: 0.907590226292854
similarity_same = spectral1 | spectral1_
print(similarity_same) # Similarity in graph spectrum: 1.0
print(spectral1.vectors.shape) # numpy array: shape=[6, 13]
print(spectral2.vectors.shape) # numpy array: shape=[6, 15]
4.Molecular assembly
The molecular assembly is handled by the standalone module
hotpot.MolAssembly temporarily.
Generic description
The molecular assembly (hotpot.cheminfo.mol_assemble) module iteratively generates virtual
molecular structures based on the user-specified molecular Framework (hotpot.Molecule) and
assembly fragments hotpot.cheminfo.mol_assemble.Fragment. The Framework is a standard
Molecule object, while the assembly operation is specifically implemented using the Fragment.
An instantiated Fragment must specify the following four factors:
- The 2D molecular structure of the fragment (a
Moleculeobject) - The atom(s) (specified by index) on the fragment used for connection with the Framework
- The searcher for locating connection sites on the Framework (a
hotpot.cheminfo.search.Searcherobject) - The specific connection operation (specified in an
action_funcfunction) between theFragmentand the Framework at the connection sites.
The Fragment provides users the flexibility to customize their own assembly strategies.
Of course, Hotpot has predefined some common molecular assembly Fragment (named Assembler).
When handling the Assembler, users only need to specify its fragment structure and indicate the
(optional) action_points indices (i.e., specify which Fragmental atoms as the "reaction site" to
react with the frame Molecule).
So far, the predefined Assembler include (see the following Scheme 1 for details):
- EdgeShoulder (required two
action_points) - AtomLink (required one
action_points) - BondAdding (No
action_pointsrequired) - AtomReplace (No
action_pointsrequired) - AlkylGraft (No
action_pointsrequired, just a specificAtomLink) - RingWedge (required one
action_points)

Scheme 1 Illustration of Assembly of Molecule by different Assemblers
5.Wet-lab experimental optimization
Hotpot also integrates a module for optimizing the wet-lab experiments using an active learning scheme. For pure parameter optimization, you can use the CLI interface:
hotpot optimize [input_excel] [output_dir] --flags args ...
hotpot optimize --help # for help
Simply follow the instructions in the command‑line interface to obtain the optimized recommended parameters.
The results and the manifold visualization of the explored parameter space are saved in output_dir.
The input_excel file should be organized as follows:
| feature1 | feature2 | ... | featureN | target |
|---|---|---|---|---|
| 0.64654 | 148.792 | ... | -30.897 | 0.3433 |
| ... | ... | ... | ... | ... |
For optimization involving molecule structures:
import numpy as np
import hotpot as hp
list_smi = [
'c1cccc1',
'c1cccc1C(=O)O',
# ...
]
mol_space = [hp.read_mol(smi) for smi in list_smi]
samples = [
hp.read_mol(list_smi[i]) for i in np.random.randint(2, size=100).tolist()
]
for mol, params in zip(samples, np.random.randn(100, 3)):
mol.add_envs(params, name=['T', 'P', 'Conc.'])
bundle = hp.MolBundle(samples)
result = bundle.optimize(
mol_space=mol_space, # Optional
env_space=..., # Optional
maximize=True, # Default
n_trails=20,
batch_size=5,
mol_repr='ComplexFormer_nano', # Optional[rdkit, fp, spectrum], The optimize method automatically selects a suitable representation.
visualize=True
)
print(result.mol.smiles)
print(result.env)
result.fig.show() # Displays the manifold visualization
🛤️ Roadmap & Project Evolution
Hotpot initially started as a more Pythonic wrapper around OpenBabel and RDKit, aiming to:
- provide a cleaner, chemist-friendly interface on the Python side, and
- avoid low-level C++ issues (e.g., segmentation faults / exit code 139) ..., and the unnatural modeling of metal complexes.
During development, it became clear that heuristic, rule-based logic is not sufficient for many real chemical problems, especially in coordination chemistry. Many chemical and biological insights are empirical and resist explicit coding.
Hotpot is therefore evolving from a rule-based wrapper into a data-driven infrastructure that tries to capture such tacit knowledge through large-scale pre-training on coordination chemistry and related databases.
Current Status
- The current
mainbranch focuses on a stable, chemist-centric core (hotpot.cheminfo.core) and classical utilities. - Several advanced AI-backed components described in this README currently live in research branches and internal prototypes, and will be merged step by step.
- Public APIs in
hotpot.cheminfo.corewill be kept as stable as possible to ensure backward compatibility as new models and pipelines are integrated.
Planned Timeline
A large part of the AI backend is closely tied to ongoing Ph.D. research work.
Major model components and pipelines are planned to be merged into the public repository progressively as the
research is completed and stabilized (target: around late 2026).
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file hotpot_zzy-0.5.2.1.tar.gz.
File metadata
- Download URL: hotpot_zzy-0.5.2.1.tar.gz
- Upload date:
- Size: 96.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.9.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5acef54e6d982afed00248bc8d1e768ef1e2739ad52d0b51a02edbe98158a65a
|
|
| MD5 |
e1bff64411bb996f0bb4651f73a7429d
|
|
| BLAKE2b-256 |
480a90a5b5a3b5e7fc982b8210af189332fd5a9b87cc5509eb0f488c07595b71
|
File details
Details for the file hotpot_zzy-0.5.2.1-py3-none-any.whl.
File metadata
- Download URL: hotpot_zzy-0.5.2.1-py3-none-any.whl
- Upload date:
- Size: 101.1 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.9.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
0ee217266c8ffa6956c464d6b140109834e6a19c136715767a07e1da81175498
|
|
| MD5 |
ee8d54b217578c663cb7e17ed06ae57b
|
|
| BLAKE2b-256 |
1c3822fb097b7d1ee2ceb4aa6243ee8b53e42a36b9c0c41d01fc66163616a2ad
|








{kind=link}