immuneML is a software platform for machine learning analysis of immune receptor repertoires.

Project description
immuneML




immuneML is a platform for machine learning-based analysis and classification of adaptive immune receptors and repertoires (AIRR).
It supports the analyses of experimental B- and T-cell receptor data, as well as synthetic data for benchmarking purposes.
In immuneML, users can define flexible workflows supporting different machine learning libraries (such as scikit-learn or PyTorch), benchmarking of different approaches, numerous reports of data characteristics, ML algorithms and their predictions, and visualizations of results.
Additionally, users can extend the platform by defining their own data representations, ML models, reports and visualizations.
Useful links:
- Main website: https://immuneml.uio.no
- Documentation: https://docs.immuneml.uio.no
- Documentation for the latest (unstable) version (development branch): https://uio-bmi.github.io/immuneML/
- Galaxy web interface (under development): https://galaxy.immuneml.uiocloud.no
Installation
We recommend installing immuneML inside a virtual environment. immuneML uses Python 3.9 or later. If using immuneML simulation, Python 3.11 or later is recommended. immuneML can be installed directly using a package manager such as pip or conda, or set up via docker.
Quick installation (immuneML essentials):
python3 -m venv ./immuneml_venv/
source ./immuneml_venv/bin/activate
pip install immuneML
or
conda create --prefix immuneml_env/ python=3.11
conda activate immuneml_env/
conda install -c bioconda immuneml
Detailed installation (immuneML extras):
Please check the documentation for more detailed instructions or how to install optional dependencies.
Validating the installation
To validate the installation, run:
immune-ml -h
This should display a help message explaining immuneML usage.
To quickly test out whether immuneML is able to run, try running the quickstart command:
immune-ml-quickstart ./quickstart_results/
This will generate a synthetic dataset and run a simple machine machine learning analysis
on the generated data. The results folder will contain two sub-folders: one for the generated dataset (synthetic_dataset)
and one for the results of the machine learning analysis (machine_learning_analysis).
The files named specs.yaml are the input files for immuneML that describe how to generate
the dataset and how to do the machine learning analysis. The index.html files can be used
to navigate through all the results that were produced.
Usage
Quickstart
The quickest way to familiarize yourself with immuneML usage is to follow one of the Quickstart tutorials. These tutorials provide a step-by-step guide on how to use immuneML for a simple machine learning analysis on an adaptive immune receptor repertoire (AIRR) dataset, using either the command line tool or the Galaxy web interface.
Overview of immuneML analyses
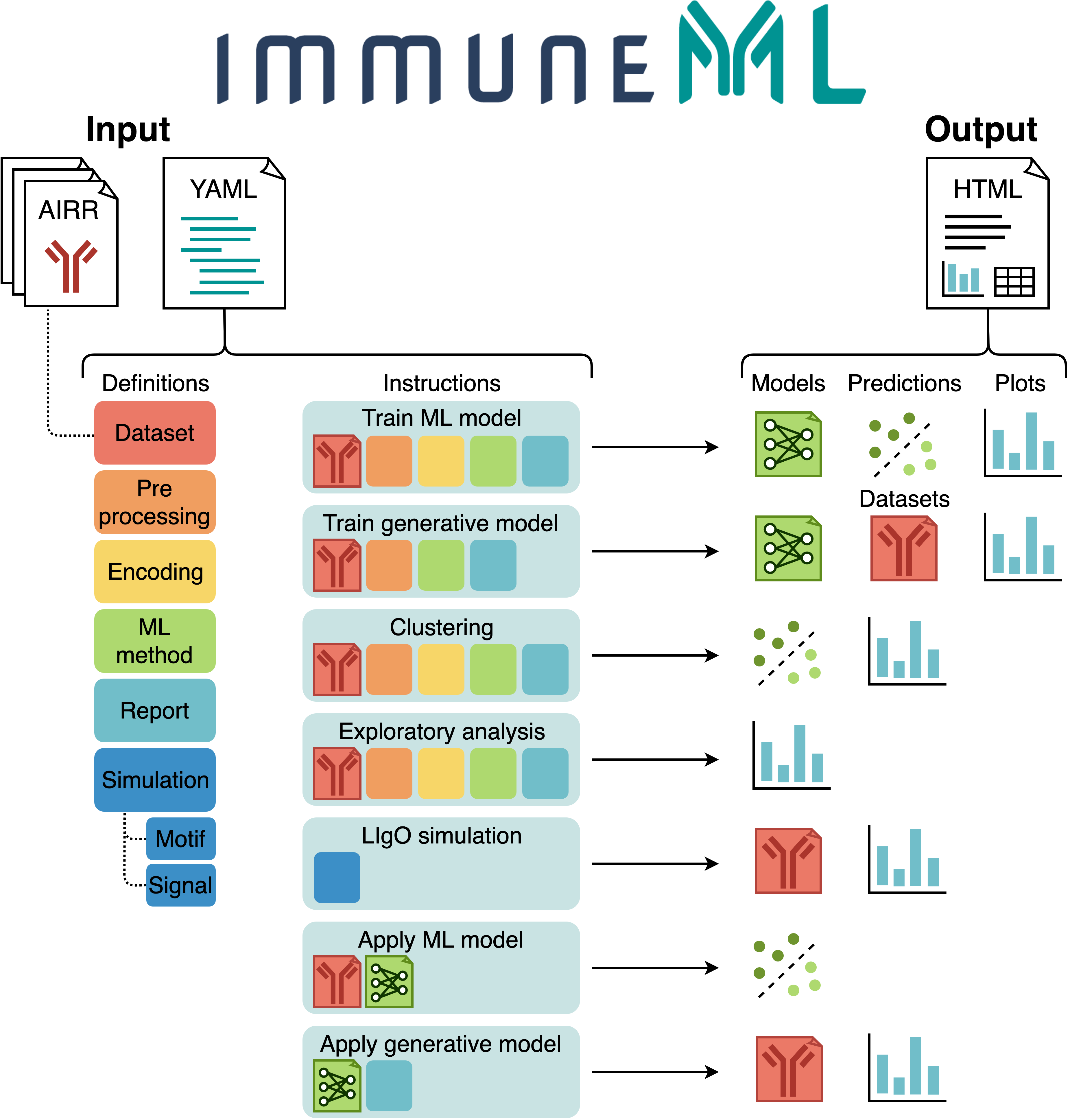
The figure below shows an overview of immuneML usage.
All parameters for an immuneML analysis are defined in the YAML specification file.
In this file, the settings of the analysis components are defined (also known as definitions,
shown in different colors in the figure).
Additionally, the YAML file describes one or more instructions, which are workflows that are
applied to the defined analysis components.
See also: documentation of the YAML specification.
Each instruction produces different types of results, including trained ML models, ML model predictions on a given dataset, plots or other reports describing the dataset or trained models, or synthetic/simulated datasets. These results can be navigated through the summary HTML file. See also: tutorials for specific immuneML use cases.
Command line usage
The immune-ml command takes only two parameters: the YAML specification file and a result path.
An example is given here:
immune-ml path/to/specification.yaml result/folder/path/
Results of an immuneML run
For each instruction specified in the YAML specification file, a subfolder is created in the
result/folder/path. Each subfolder will contain:
- An
index.htmlfile which shows an overview of the results produced by that instruction. Inspecting the results of an immuneML analysis typically starts here. - A copy of the used YAML specification (
full_specification.yaml) with all default parameters explicitly set. - A log file (
log.txt). - A folder containing the imported dataset(s) in immuneML format.
- A folder containing all raw results produced by the instruction.
Support
We will prioritize fixing important bugs, and try to answer any questions as soon as possible. Please note we are only 2 people maintaining the platform with occasional absences.
When experiencing an issue, please take the following steps:
-
Make sure the latest version of immuneML is installed. immuneML is under constant development, and the issue you experience may already be resolved in the latest version of the platform.
-
Check the 'troubleshooting' page in the immuneML documentation. Any known issues and their solutions are already described there.
-
If you are still experience a problem and suspect a bug in immuneMl, you can report an issue on GitHub. Please make sure to include the following information:
- The YAML specification you tried to run.
- The full output log file (log.txt).
- A list of dependency versions (can be retrieved with pip list or conda list).
- We primarily test immuneML using Unix-based operating systems, please make sure to mention it if you're using Windows.
- We will be able to help you fastest if you can also provide a small reproducible example, such as a very small dataset for which your run fails.
If this does not answer your question, you can contact us via:
- Twitter
@immuneml - Email
contact@immuneml.uio.no
Citing immuneML
If you are using immuneML in any published work, please cite:
Pavlović, M., Scheffer, L., Motwani, K. et al. The immuneML ecosystem for machine learning analysis of adaptive immune receptor repertoires. Nat Mach Intell 3, 936–944 (2021). https://doi.org/10.1038/s42256-021-00413-z
© Copyright 2021-2022, Milena Pavlovic, Lonneke Scheffer, Keshav Motwani, Victor Greiff, Geir Kjetil Sandve
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file immuneml-3.0.27.tar.gz.
File metadata
- Download URL: immuneml-3.0.27.tar.gz
- Upload date:
- Size: 582.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d8fc2b67b9d178630b9023f7c2f5f5e060153956c843a373e9bf3f362f333ce1
|
|
| MD5 |
1400e3e8057fe9c33c7b493848dd11a8
|
|
| BLAKE2b-256 |
d5be9c1192fff973217b214d7197d09bfa349416cbb27d39bb389f5b839919b2
|
File details
Details for the file immuneml-3.0.27-py3-none-any.whl.
File metadata
- Download URL: immuneml-3.0.27-py3-none-any.whl
- Upload date:
- Size: 900.0 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d10dad6f25c008788e9af9313a95a24c8237c666cbc1ac4cfe586b4c17d85fd3
|
|
| MD5 |
c1ec923b16ff9b4589c7acf545a7b7f2
|
|
| BLAKE2b-256 |
6e9a87338e7ac12bd894e1718d1bd885e4599cfbbb2e6761cd4589dc1fb1f8d4
|







