Python utility libraries on genome assembly, annotation and comparative genomics

Project description
JCVI: A Versatile Toolkit for Comparative Genomics Analysis




Collection of Python libraries to parse bioinformatics files, or perform computation related to assembly, annotation, and comparative genomics.
| Authors | Haibao Tang (tanghaibao) |
| Vivek Krishnakumar (vivekkrish) | |
| Adam Taranto (Adamtaranto) | |
| Xingtan Zhang (tangerzhang) | |
| Won Cheol Yim (wyim-pgl) | |
| tanghaibao@gmail.com | |
| License | BSD |
How to cite
[!TIP] JCVI is now published in iMeta!
Tang et al. (2024) JCVI: A Versatile Toolkit for Comparative Genomics Analysis. iMeta
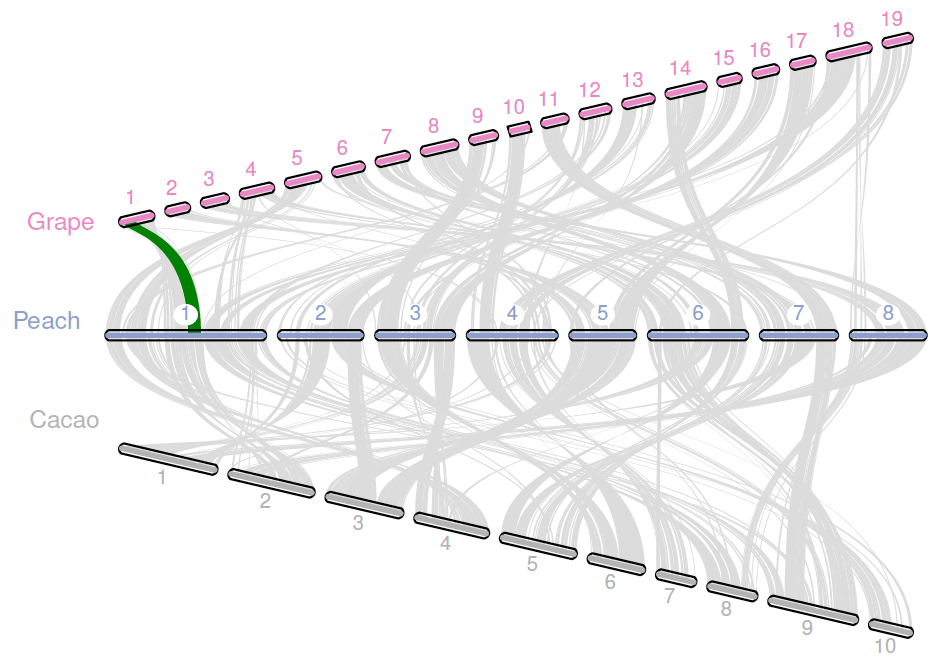
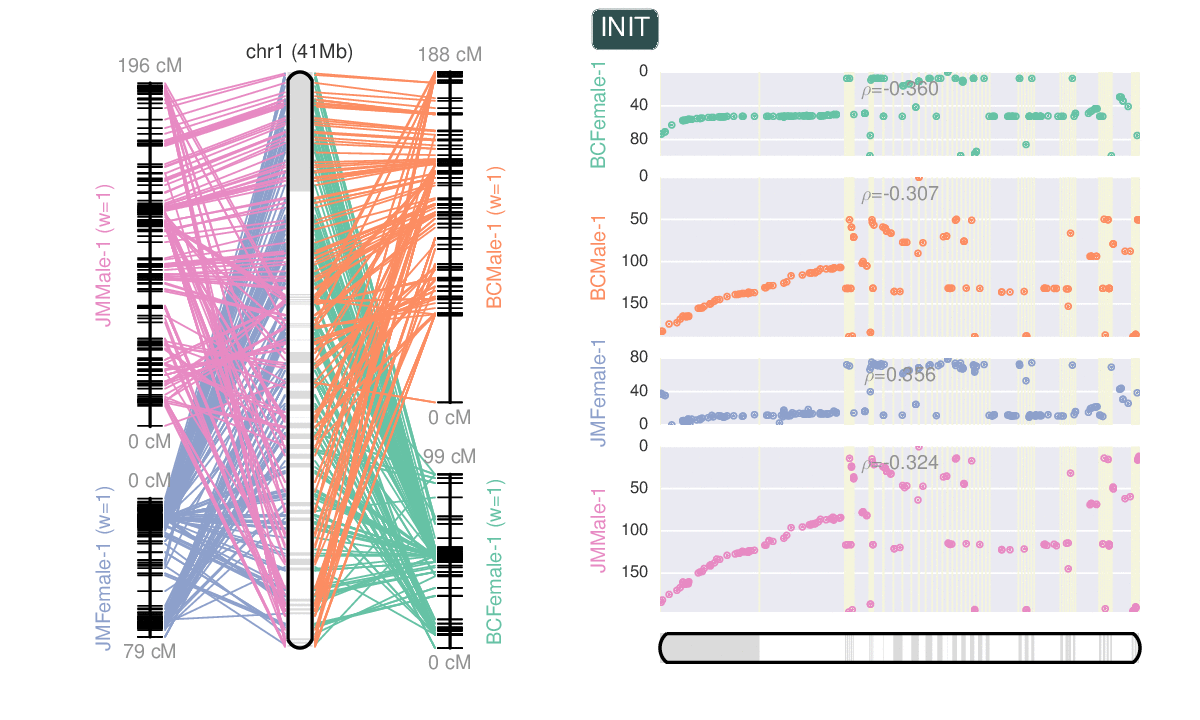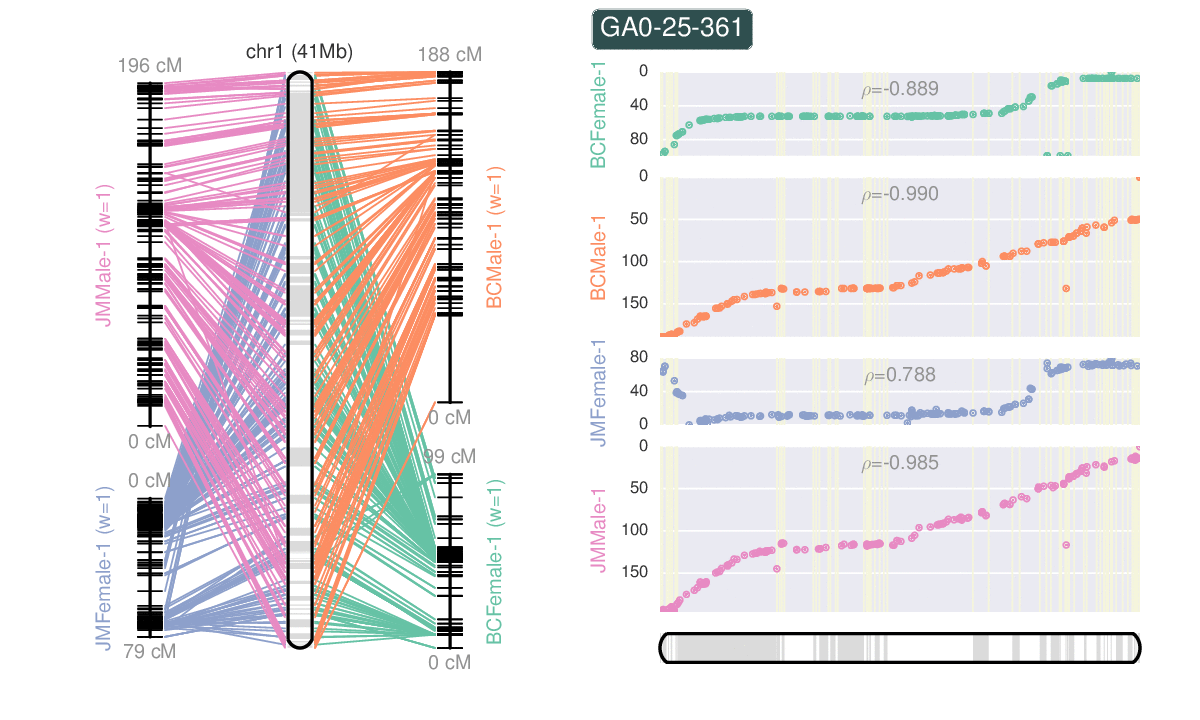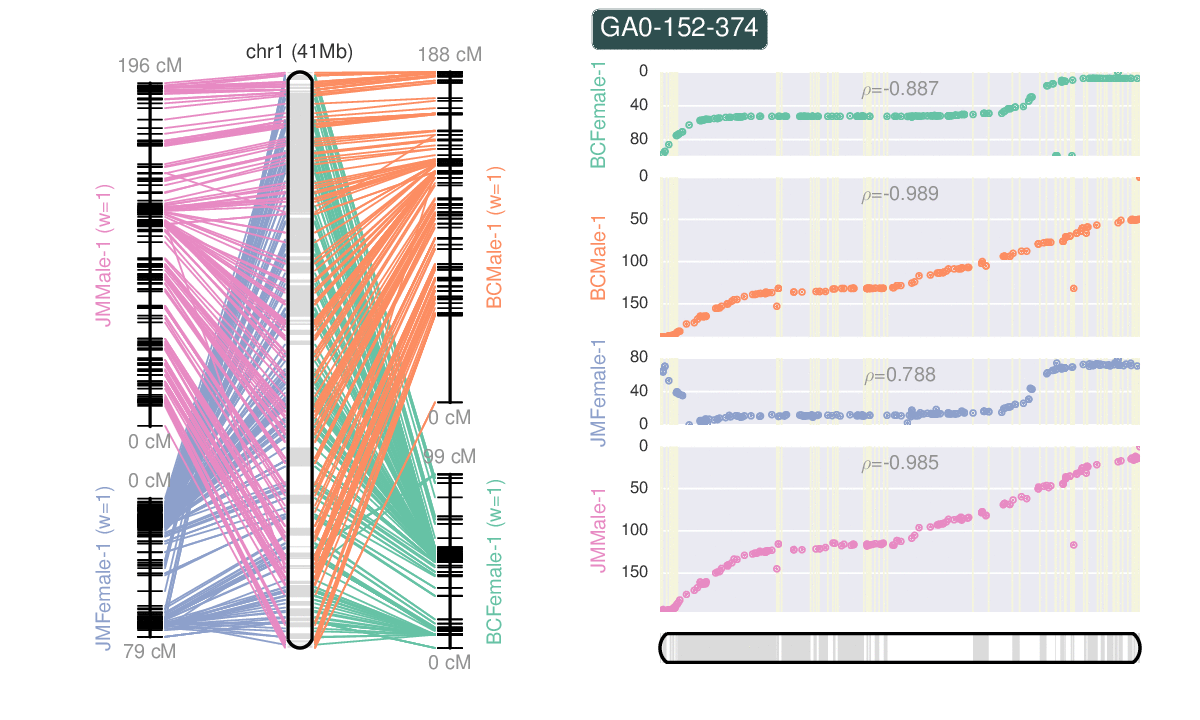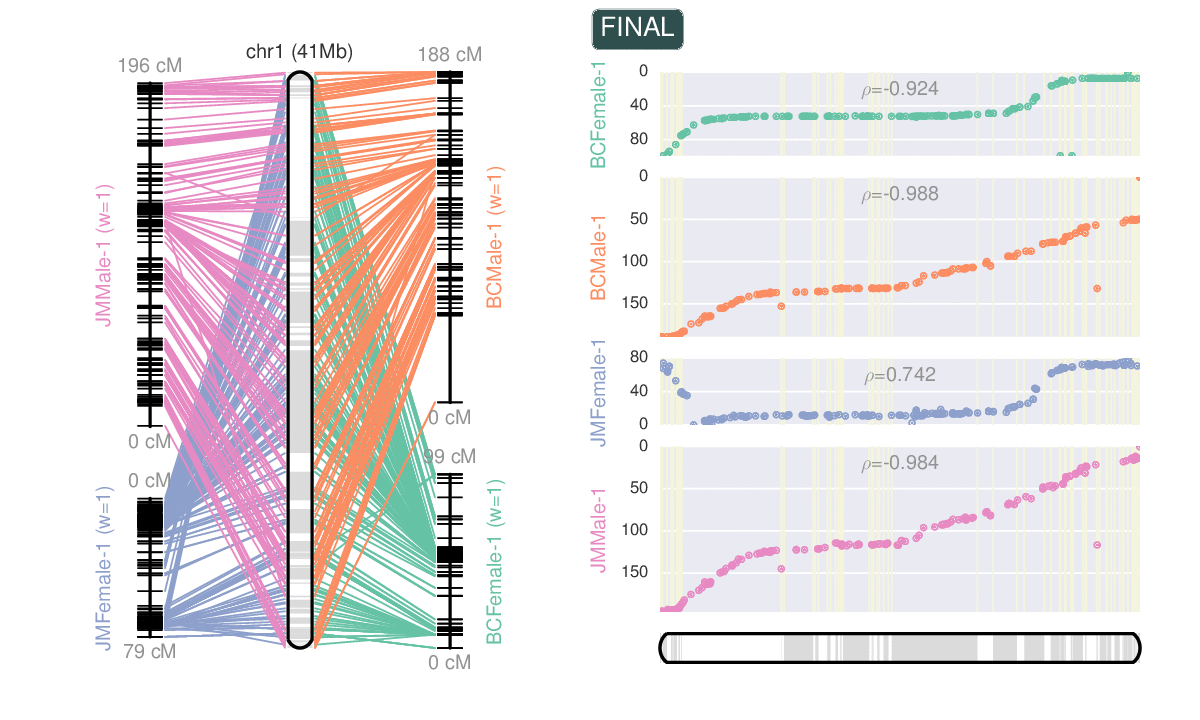
Contents
Following modules are available as generic Bioinformatics handling methods.
-
algorithms
- Linear programming solver with SCIP and GLPK.
- Supermap: find set of non-overlapping anchors in BLAST or NUCMER output.
- Longest or heaviest increasing subsequence.
- Matrix operations.
-
apps
- GenBank entrez accession, Phytozome, Ensembl and SRA downloader.
- Calculate (non)synonymous substitution rate between gene pairs.
- Basic phylogenetic tree construction using PHYLIP, PhyML, or RAxML, and viualization.
- Wrapper for BLAST+, LASTZ, LAST, BWA, BOWTIE2, CLC, CDHIT, CAP3, etc.
-
formats
Currently supports
.aceformat (phrap, cap3, etc.),.agp(goldenpath),.bedformat,.blastoutput,.btabformat,.coordsformat (nucmeroutput),.fastaformat,.fastqformat,.fpcformat,.gffformat,oboformat (ontology),.pslformat (UCSC blat, GMAP, etc.),.posmapformat (Celera assembler output),.samformat (read mapping),.contigformat (TIGR assembly format), etc. -
graphics
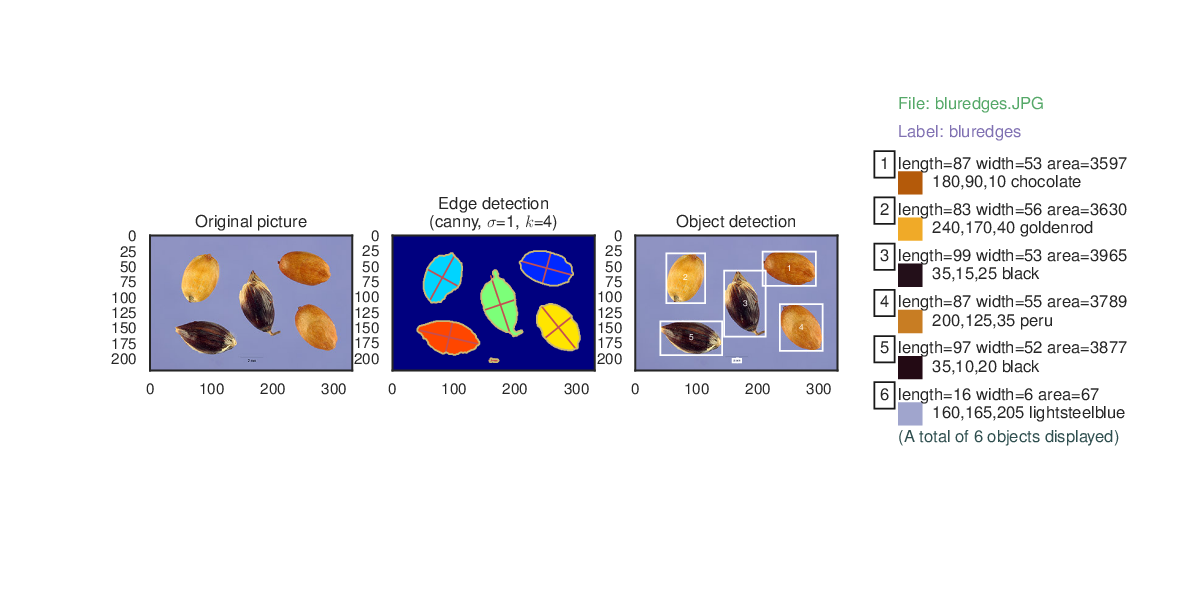
- BLAST or synteny dot plot.
- Histogram using R and ASCII art.
- Paint regions on set of chromosomes.
- Macro-synteny and micro-synteny plots.
- Ribbon plots from whole genome alignments.
-
utils
- Grouper can be used as disjoint set data structure.
- range contains common range operations, like overlap and chaining.
- Miscellaneous cookbook recipes, iterators decorators, table utilities.
Then there are modules that contain domain-specific methods.
-
assembly
- K-mer histogram analysis.
- Preparation and validation of tiling path for clone-based assemblies.
- Scaffolding through ALLMAPS, optical map and genetic map.
- Pre-assembly and post-assembly QC procedures.
-
annotation
- Training of ab initio gene predictors.
- Calculate gene, exon and intron statistics.
- Wrapper for PASA and EVM.
- Launch multiple MAKER processes.
-
compara
- C-score based BLAST filter.
- Synteny scan (de-novo) and lift over (find nearby anchors).
- Ancestral genome reconstruction using Sankoff's and PAR method.
- Ortholog and tandem gene duplicates finder.
Applications
Please visit wiki for full-fledged applications.
Dependencies
JCVI requires Python3 between v3.9 and v3.12.
Some graphics modules require the ImageMagick library.
On MacOS this can be installed using Conda (see next section). If you are using a linux system (i.e. Ubuntu) you can install ImageMagick using apt-get:
sudo apt-get update
sudo apt-get install libmagickwand-dev
See the Wand docs for instructions on installing ImageMagick on other systems.
A few modules may ask for locations of external programs,
if the executable cannot be found in your PATH.
The external programs that are often used are:
Managing dependencies with Conda
You can use the the YAML files in this repo to create an environment with basic JCVI dependencies.
If you are new to Conda, we recommend the Miniforge distribution.
conda env create -f environment.yml
conda activate jcvi
After activating the Conda environment install JCVI using one of the following options.
Installation
Installation options
- Use pip to install the latest development version directly from this repo.
pip install git+https://github.com/tanghaibao/jcvi.git
- Install latest release from PyPi.
pip install jcvi
- Alternatively, if you want to install in development mode.
git clone git://github.com/tanghaibao/jcvi.git && cd jcvi
pip install -e '.[tests]'
pre-commit install
Test Installation
If installed successfully, you can check the version with:
jcvi --version
Usage
Use python -m to call any of the modules installed with JCVI.
Most of the modules in this package contains multiple actions. To use
the fasta example:
Usage:
python -m jcvi.formats.fasta ACTION
Available ACTIONs:
clean | Remove irregular chars in FASTA seqs
diff | Check if two fasta records contain same information
extract | Given fasta file and seq id, retrieve the sequence in fasta format
fastq | Combine fasta and qual to create fastq file
filter | Filter the records by size
format | Trim accession id to the first space or switch id based on 2-column mapping file
fromtab | Convert 2-column sequence file to FASTA format
gaps | Print out a list of gap sizes within sequences
gc | Plot G+C content distribution
identical | Given 2 fasta files, find all exactly identical records
ids | Generate a list of headers
info | Run `sequence_info` on fasta files
ispcr | Reformat paired primers into isPcr query format
join | Concatenate a list of seqs and add gaps in between
longestorf | Find longest orf for CDS fasta
pair | Sort paired reads to .pairs, rest to .fragments
pairinplace | Starting from fragment.fasta, find if adjacent records can form pairs
pool | Pool a bunch of fastafiles together and add prefix
qual | Generate dummy .qual file based on FASTA file
random | Randomly take some records
sequin | Generate a gapped fasta file for sequin submission
simulate | Simulate random fasta file for testing
some | Include or exclude a list of records (also performs on .qual file if available)
sort | Sort the records by IDs, sizes, etc.
summary | Report the real no of bases and N's in fasta files
tidy | Normalize gap sizes and remove small components in fasta
translate | Translate CDS to proteins
trim | Given a cross_match screened fasta, trim the sequence
trimsplit | Split sequences at lower-cased letters
uniq | Remove records that are the same
Then you need to use one action, you can just do:
python -m jcvi.formats.fasta extract
This will tell you the options and arguments it expects.
Feel free to check out other scripts in the package, it is not just for FASTA.
Star History

Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file jcvi-1.5.11.tar.gz.
File metadata
- Download URL: jcvi-1.5.11.tar.gz
- Upload date:
- Size: 789.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ea6ce01316fa52e7d53ea6d8e7bf6da840f2314996cba3283e4b20438786716f
|
|
| MD5 |
9f8508f926e2e57c0d6f7d34997f4f8d
|
|
| BLAKE2b-256 |
57d497a5c1f03ab9dd3f2ee82b071010c7fac443cbf346a76eeda9c847991de0
|
File details
Details for the file jcvi-1.5.11-cp312-cp312-macosx_11_0_arm64.whl.
File metadata
- Download URL: jcvi-1.5.11-cp312-cp312-macosx_11_0_arm64.whl
- Upload date:
- Size: 1.2 MB
- Tags: CPython 3.12, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
58572f6fe995449f89c718e2cea3a22b8810a7ba648430f7c6175366a9e748b5
|
|
| MD5 |
42b1785e09cf3e2d390cb5f690a843ef
|
|
| BLAKE2b-256 |
d23b73895b439a71e0fccb96db9e002ef7ab4851d44da30d1797a112541dc9aa
|







