No project description provided

Project description
JIMG_ncd – Python library for automated nucleus detection and analysis





Author: Jakub Kubiś
Polish Academy of Sciences
Description
JIMG_ncd is a Python library designed for DL based automated nucleus detection and analysis in high-resolution images from confocal microscopy and flow cytometry, eg. Amnis-ImageStream. It facilitates in-depth examination of nuclei and their chromatin organization, enabling precise analysis of nuclear morphology and chromatin structure.
Additionally, JIMG_ncd provides advanced tools for analyzing cell populations from cytometric data. Users can select distinctive cellular features, cluster cells based on these characteristics, and apply statistical analyses to uncover unique features within each cluster.
📚 Table of Contents
- 1.Installation
- 2.Documentation
- 3.Example pipelines
-
3.1. Nuclei analysis - confocal microscopy
- 3.1.1 Testing analysis parameters
- 3.1.2 Performing nuclei analysis with adjusted parameters
- 3.1.3 Selecting nuclei based on nucleus parameters
- 3.1.4 Extracting nuclei chromanitization features
- 3.1.5 Adjusting nuclei chromanitization parameters
- 3.1.6 Analyzing nuclei series
- 3.1.7 Obtaining nuclei series analysis results
- 3.1.8 Analyzing nuclei chromatinization series
- 3.1.9 Obtaining nuclei chromatinization series analysis results
-
3.2. Nuclei analysis - flow cytometry
- 3.2.1 Testing analysis parameters
- 3.2.2 Performing nuclei analysis with adjusted parameters
- 3.2.3 Selecting nuclei based on nucleus parameters
- 3.2.4 Extracting nuclei chromanitization features
- 3.2.5 Adjusting nuclei chromanitization parameters
- 3.2.6 Analyzing nuclei chromatinization series
- 3.2.7 Obtaining nuclei chromatinization series analysis results
- 3.2.8 Concatenating nuclei chromatinization analysis with ImageStream (IS) data
- 3.2.9 Combining projects from nuclei chromatinization analysi
-
3.3. Clustering and DFA (Differential Feature Analysis) – nuclei data
- 3.3.1 Selecting feature analysis (DFA) for separate experiments data
- 3.3.2 Loading images IDs
- 3.3.3 Performing data scaling and dimensionality reduction
- 3.3.4 Performing UMAP & clustering
- 3.3.5 Obtaining complete data and metadata (clusters)
- 3.3.6 Preforming DFA analysis on clusters
- 3.3.7 Preforming proportion analysis
-
1. Installation
In command line write:
pip install jimg-ncd
2. Documenation
Documentation for classes and functions is available here 👉 Documentation 📄
3. Example pipelines
If you want to run the examples, you must download the test data. To do this, use:
from jimg_ncd.nuclei import test_data
test_data()
3.1 Nuclei analysis - confocal microscopy
3.1.1 Testing analysis parameters
from jimg_ncd.nuclei import NucleiFinder
# initiate class
nf = NucleiFinder()
image = nf.load_image('test_data/microscope_nuclei/r01c02f90p20-ch1sk1fk1fl1.tiff')
nf.input_image(image)
# Check the basic parameters
nf.current_parameters_nuclei
# Test nms & prob parmeters for nuclei segmentation
nf.nuclei_finder_test()
nf.browser_test()
# If required, change parameters
nf.set_nms(nms = 0.9)
nf.set_prob(prob = 0.5)
# Analysis
# 1. First step on nuclei analysis
nf.find_nuclei()
# Parameters for micrsocope image adjustment
nf.current_parameters_img_adj
Image with 'Default' parameters:
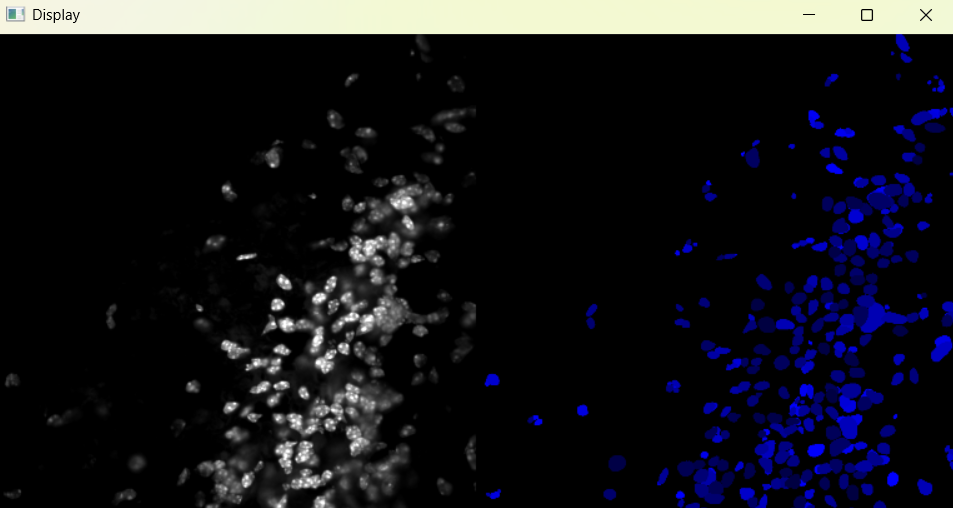
# If image required changes, change parameters and run again (nf.find_nuclei())
nf.set_adj_image_brightness(brightness = 1000)
nf.set_adj_image_gamma(gamma = 1.2)
nf.set_adj_image_contrast(contrast = 2)
# Check if parameters has changed
nf.current_parameters_nuclei
# Second execution with new parameters for image adjustment
nf.find_nuclei()
Image with adjusted parameters:
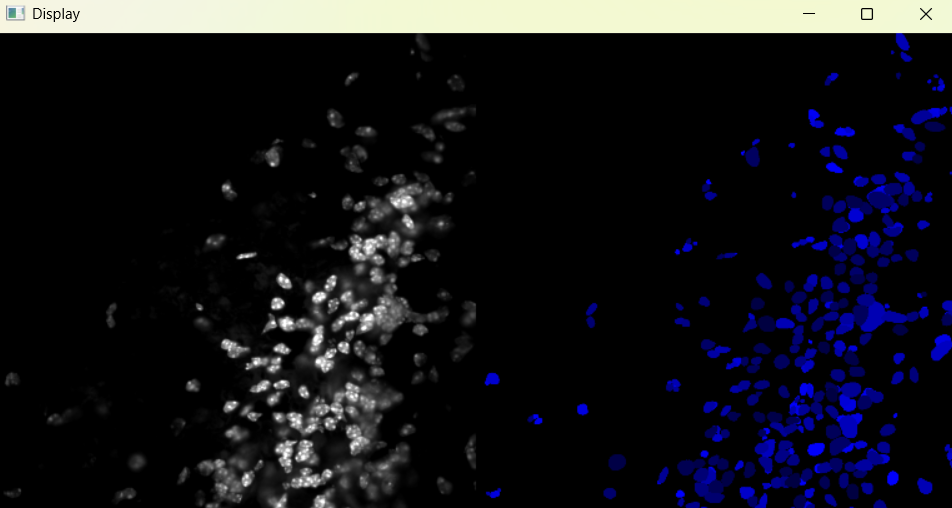
3.1.2 Performing nuclei analysis with adjusted parameters
# Return results
nf.find_nuclei()
nuclei_results, analysed_img = nf.get_results_nuclei()
Dictionary with nuclei results:
3.1.3 Selecting nuclei based on nucleus parameters
# 2. Second step of analysis (selection)
nf.select_nuclei()
Image with 'Default' selection parameters:
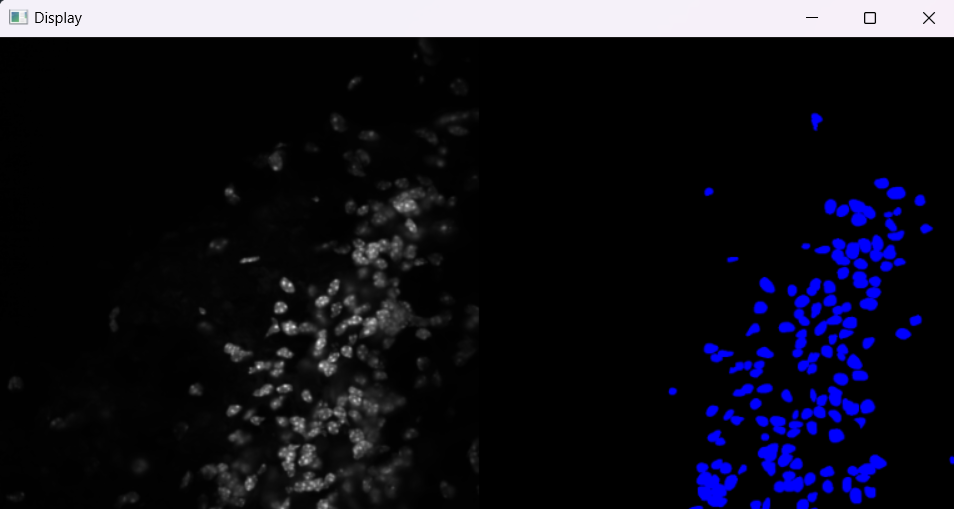
# Parameters for selecting nuclei; adjust if analysis results do not meet
# requirements, and re-run the analysis as needed.
nf.current_parameters_nuclei
nf.set_nuclei_circularity(circ = 0.5)
nf.set_nuclei_size(size = (100,800))
nf.set_nuclei_min_mean_intensity(intensity = 2000)
# Check if parameters has changed
nf.current_parameters_nuclei
# Second execution with adjusted parameters of second step of analysis (selection)
nf.select_nuclei()
Image with adjusted selection parameters:
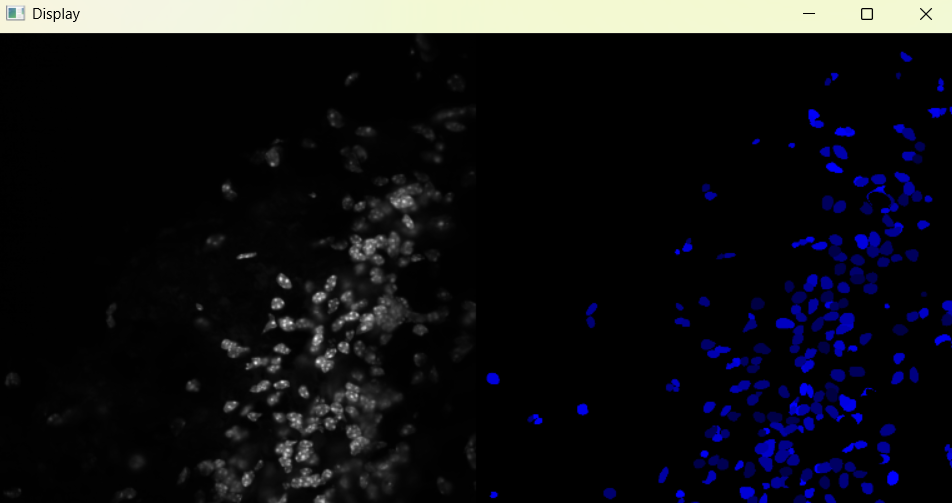
# Return results
nuclei_selected_results, analysed_selected_img = nf.get_results_nuclei_selected()
Dictionary with nuclei results:
3.1.4 Extracting nuclei chromanitization features
# 3. third step (chromatinization alaysis)
nf.nuclei_chromatinization()
Image with 'Default' chromatinization parameters:
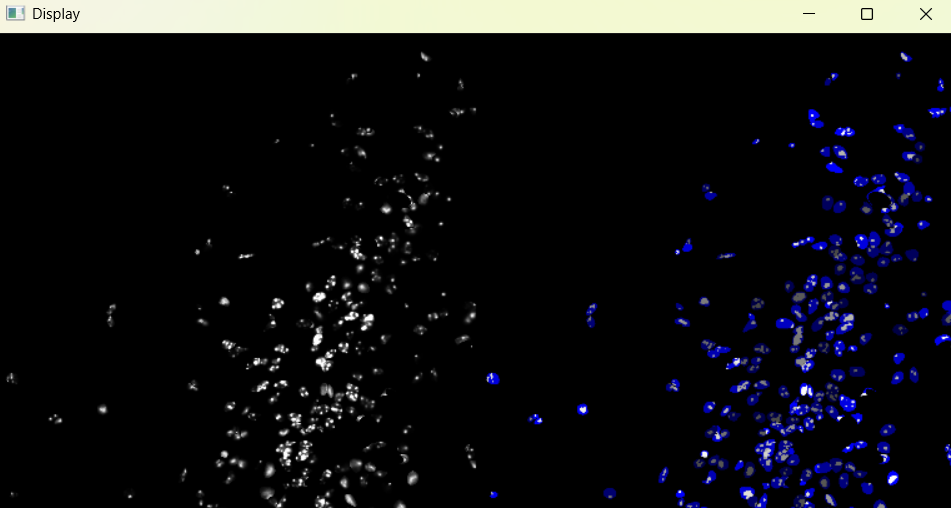
3.1.5 Adjusting nuclei chromanitization parameters
# Parameters for nuclei chromatinization; adjust if analysis results do not meet
# requirements, and re-run the analysis as needed.
# Chromatinization parameters
nf.current_parameters_chromatinization
nf.set_chromatinization_size(size = (2,400))
nf.set_chromatinization_ratio(ratio = .05)
nf.set_chromatinization_cut_point(cut_point = .95)
nf.current_parameters_chromatinization
# Chromatinization image parameters
nf.current_parameters_img_adj_chro
nf.set_adj_chrom_gamma(gamma = 0.25)
nf.set_adj_chrom_contrast(contrast = 3)
nf.set_adj_chrom_brightness(brightness = 950)
nf.current_parameters_img_adj_chro
# Second execution of the third step (chromatinization analysis)
nf.nuclei_chromatinization()
Image with adjusted chromatinization parameters:
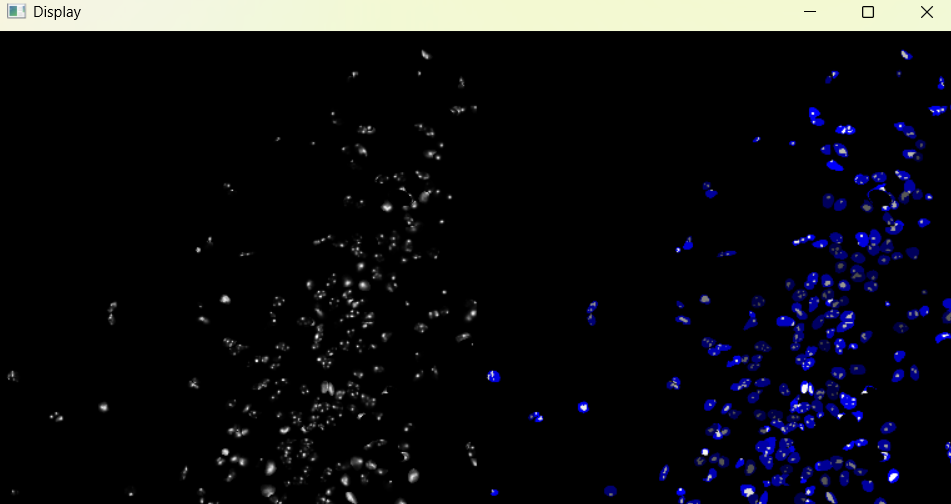
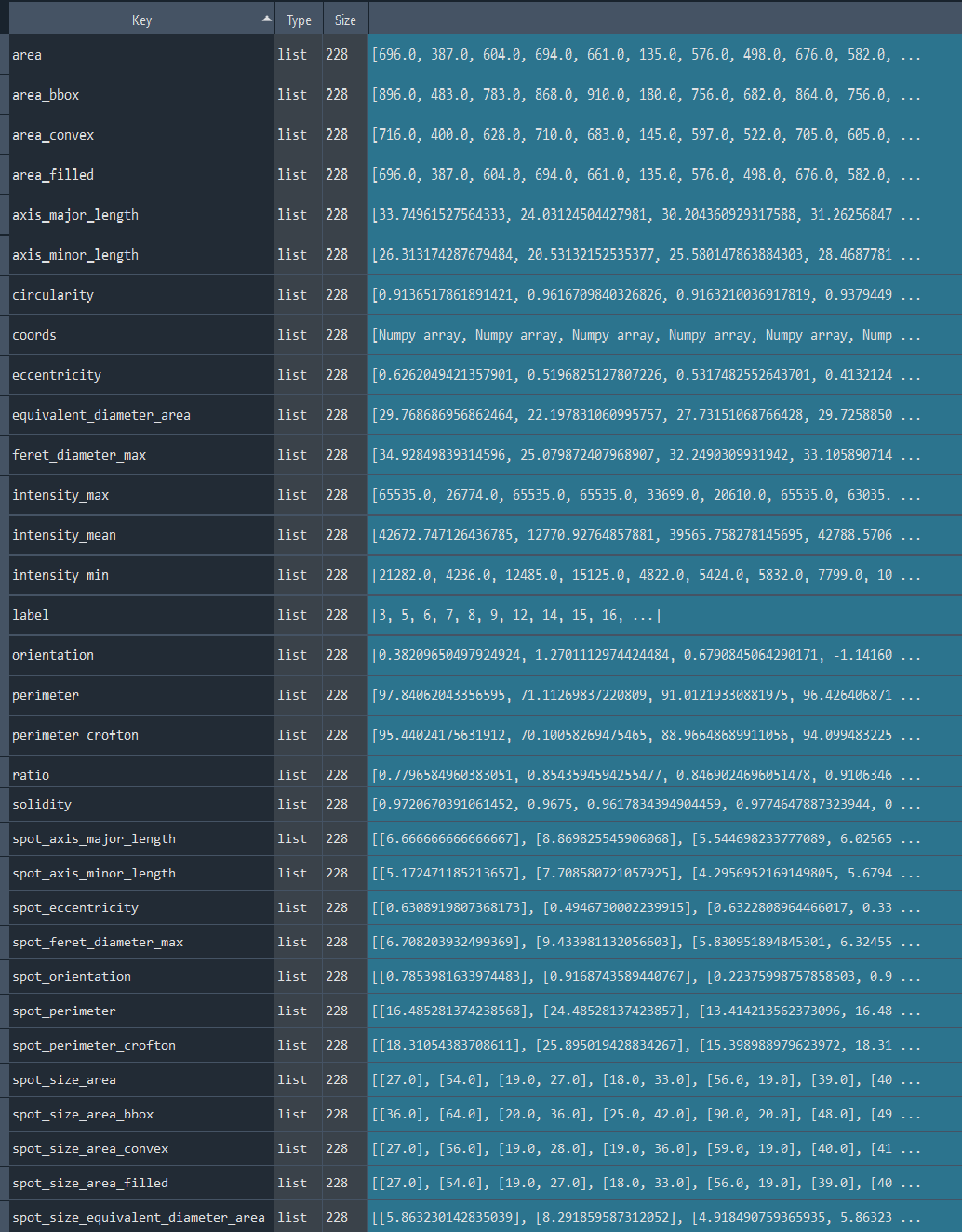
chromatinization_results, analysed_chromatinization_img = nf.get_results_nuclei_chromatinization()
Dictionary with nuclei chromatinization results:
3.1.6 Analyzing nuclei series
# If your parameters are correct for your data, you can run series analysis on more images
# Nuclei
series_results_nuclei = nf.series_analysis_nuclei(path_to_images = 'test_data/microscope_nuclei',
file_extension = 'tiff',
selected_id = [],
fille_name_part = 'ch1',
selection_opt = True,
include_img = False,
test_series = 0)
Dictionary with series nuclei results:

3.1.7 Obtaining nuclei series analysis results
# get & save results
from jimg_ncd.nuclei import NucleiDataManagement
# initiate class with NucleiFinder data
ndm = NucleiDataManagement(series_results_nuclei, 'example')
# get data as data frame
df = ndm.get_data()
print(df)
# save results as data frame
ndm.save_results_df(path='')
# save results as project *.nuc
ndm.save_nuc_project(path='')
# saved project by ndm.save_nuc_project(path='') can be then loaded for further analysis
ndm2 = NucleiDataManagement.load_nuc_dict('example.nuc')
ndm2.get_data()
3.1.8 Analyzing nuclei chromatinization series
# Chromatinization
series_results_chromatinization = nf.series_analysis_chromatinization(path_to_images = 'test_data/microscope_nuclei',
file_extension = 'tiff',
selected_id = [],
fille_name_part = 'ch1',
selection_opt = True,
include_img = True,
test_series = 0)
Dictionary with series nuclei chromatinization results:
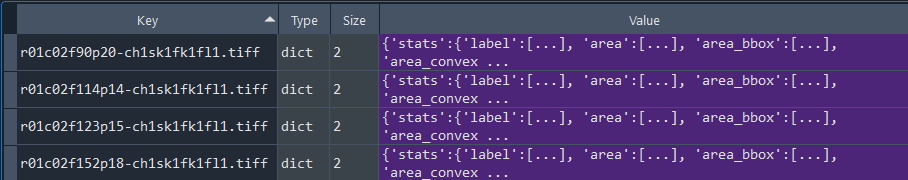
3.1.9 Obtaining nuclei chromatinization series analysis results
# get & save results
from jimg_ncd.nuclei import NucleiDataManagement
# initiate class with NucleiFinder data
ndm = NucleiDataManagement(series_results_chromatinization, 'example_chromatinization')
# get data as data frame
df = ndm.get_data()
print(df)
# save results as data frame
ndm.save_results_df(path='')
# save results as project *.nuc
ndm.save_nuc_project(path='')
# saved project by ndm.save_nuc_project(path='') can be then loaded for further analysis
ndm2 = NucleiDataManagement.load_nuc_dict('example_chromatinization.nuc')
ndm2.get_data()
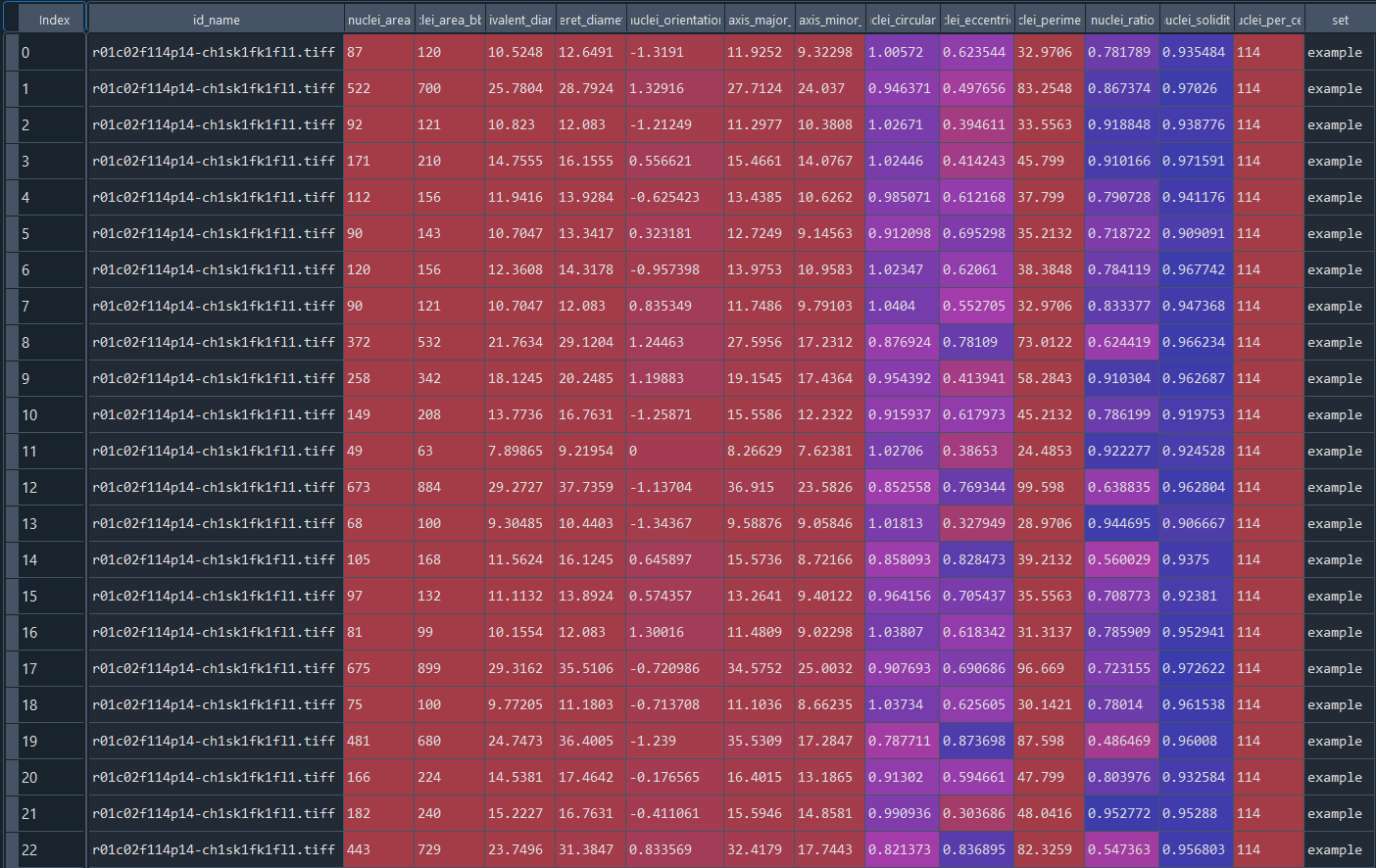
Data table with series nuclei chromatinization results:
3.2 Nuclei analysis - flow cytometry
3.2.1 Testing analysis parameters
from jimg_ncd.nuclei import NucleiFinder
# initiate class
nf = NucleiFinder()
image = nf.load_image("test_data/flow_cytometry/ctrl/3087_Ch7.ome.tif")
nf.input_image(image)
# Check the basic parameters
nf.current_parameters_nuclei
# Test nms & prob parmeters for nuclei segmentation
nf.nuclei_finder_test()
nf.browser_test()
# If required, change parameters
nf.set_nms(nms = 0.6)
nf.set_prob(prob = 0.3)
# Analysis
# 1. First step on nuclei analysis
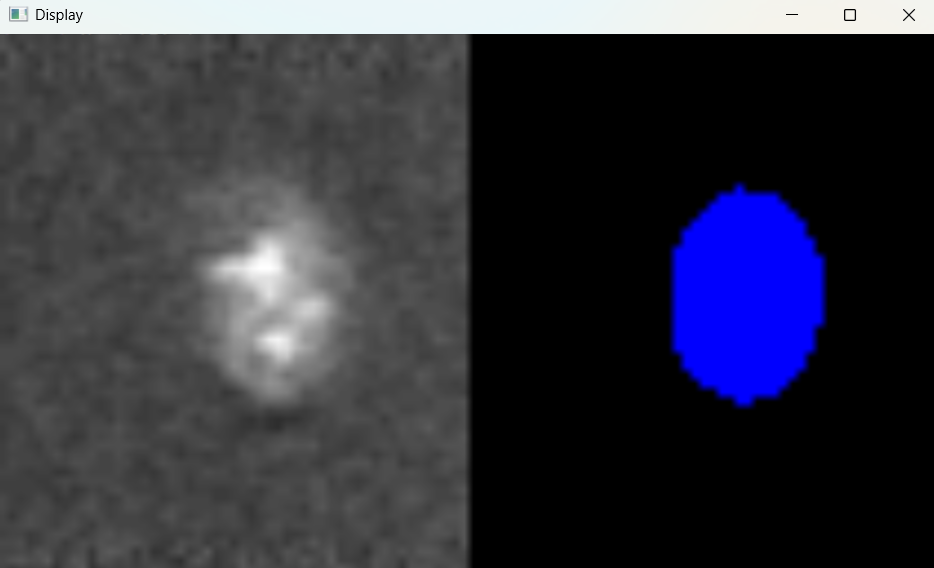
nf.find_nuclei()
Image with 'Default' parameters:
# Parameters for micrsocope image adjustment
nf.current_parameters_img_adj
# If image required changes, change parameters and run again (nf.find_nuclei())
nf.set_adj_image_brightness(brightness = 1000)
nf.set_adj_image_gamma(gamma = 1.2)
nf.set_adj_image_contrast(contrast = 2)
# Check if parameters has changed
nf.current_parameters_nuclei
# Second execution with new parameters for image adjustment
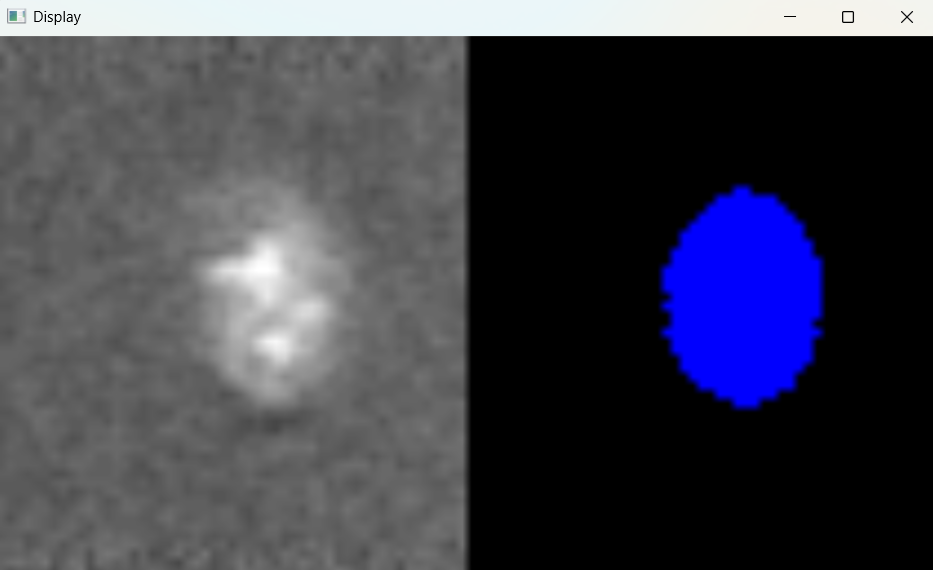
nf.find_nuclei()
Image with adjusted parameters:
3.2.2 Performing nuclei analysis with adjusted parameters
# Return results
nf.find_nuclei()
nuclei_results, analysed_img = nf.get_results_nuclei()
Dictionary with nuclei results:
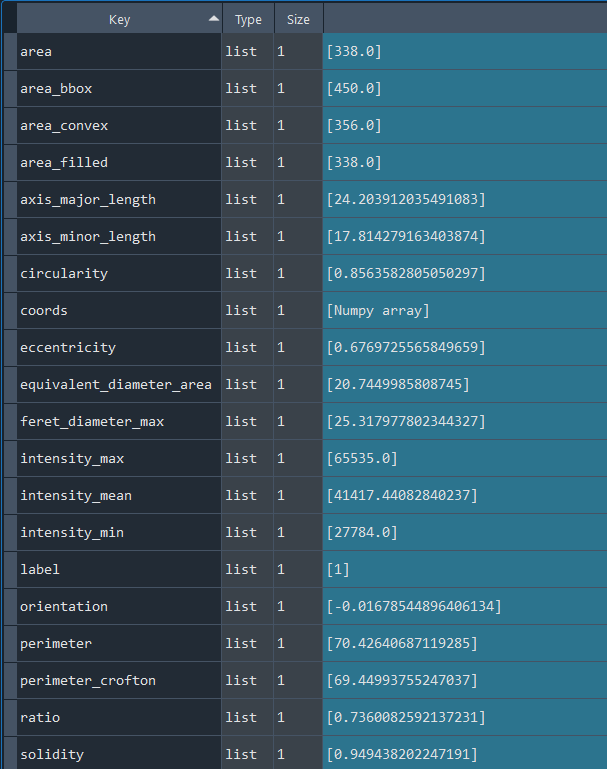
3.2.3 Selecting nuclei based on nucleus parameters
# 2. Second step of analysis (selection)
nf.select_nuclei()
Image with 'Default' selection parameters:

# Parameters for selecting nuclei; adjust if analysis results do not meet
# requirements, and re-run the analysis as needed.
nf.current_parameters_nuclei
nf.set_nuclei_circularity(circ = 0.5)
nf.set_nuclei_size(size = (100,800))
nf.set_nuclei_min_mean_intensity(intensity = 2000)
# Check if parameters has changed
nf.current_parameters_nuclei
# Second execution with adjusted parameters of second step of analysis (selection)
nf.select_nuclei()
Image with adjusted selection parameters:
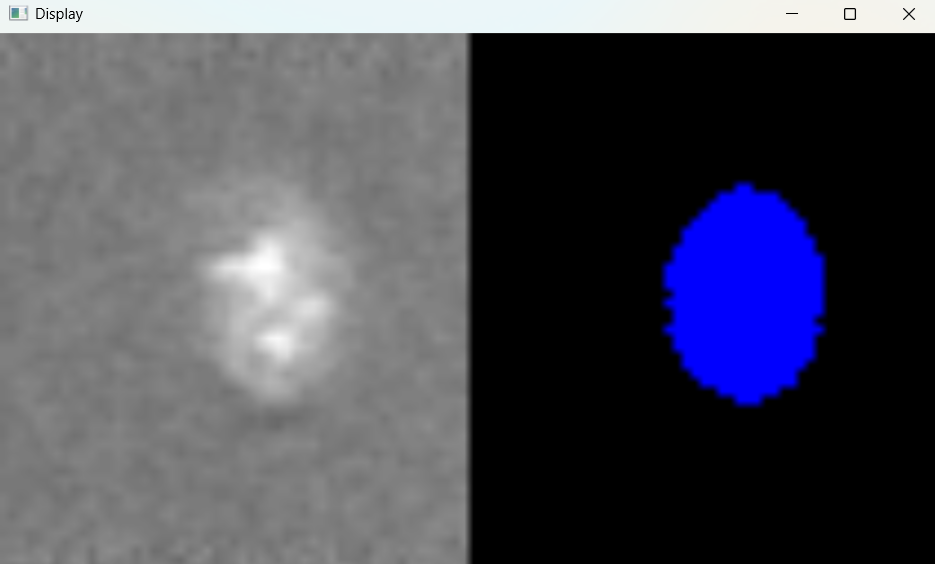
# Return results
nuclei_selected_results, analysed_selected_img = nf.get_results_nuclei_selected()
Dictionary with nuclei results:
3.2.4 Extracting nuclei chromanitization features
# 3. third step (chromatinization alaysis)
nf.nuclei_chromatinization()
Image with 'Default' chromatinization parameters:

3.2.5 Adjusting nuclei chromanitization parameters
# Parameters for nuclei chromatinization; adjust if analysis results do not meet
# requirements, and re-run the analysis as needed.
# Chromatinization parameters
nf.current_parameters_chromatinization
nf.set_chromatinization_size(size = (2,1000))
nf.set_chromatinization_ratio(ratio = 0.005)
nf.set_chromatinization_cut_point(cut_point = 1.05)
nf.current_parameters_chromatinization
# Chromatinization image parameters
nf.current_parameters_img_adj_chro
nf.set_adj_chrom_gamma(gamma = 0.25)
nf.set_adj_chrom_contrast(contrast = 4)
nf.set_adj_chrom_brightness(brightness = 950)
nf.current_parameters_img_adj_chro
# Second execution of the third step (chromatinization analysis)
nf.nuclei_chromatinization()
Image with adjusted chromatinization parameters:

# Return results
chromatinization_results, analysed_chromatinization_img = nf.get_results_nuclei_chromatinization()
Dictionary with nuclei chromatinization results:
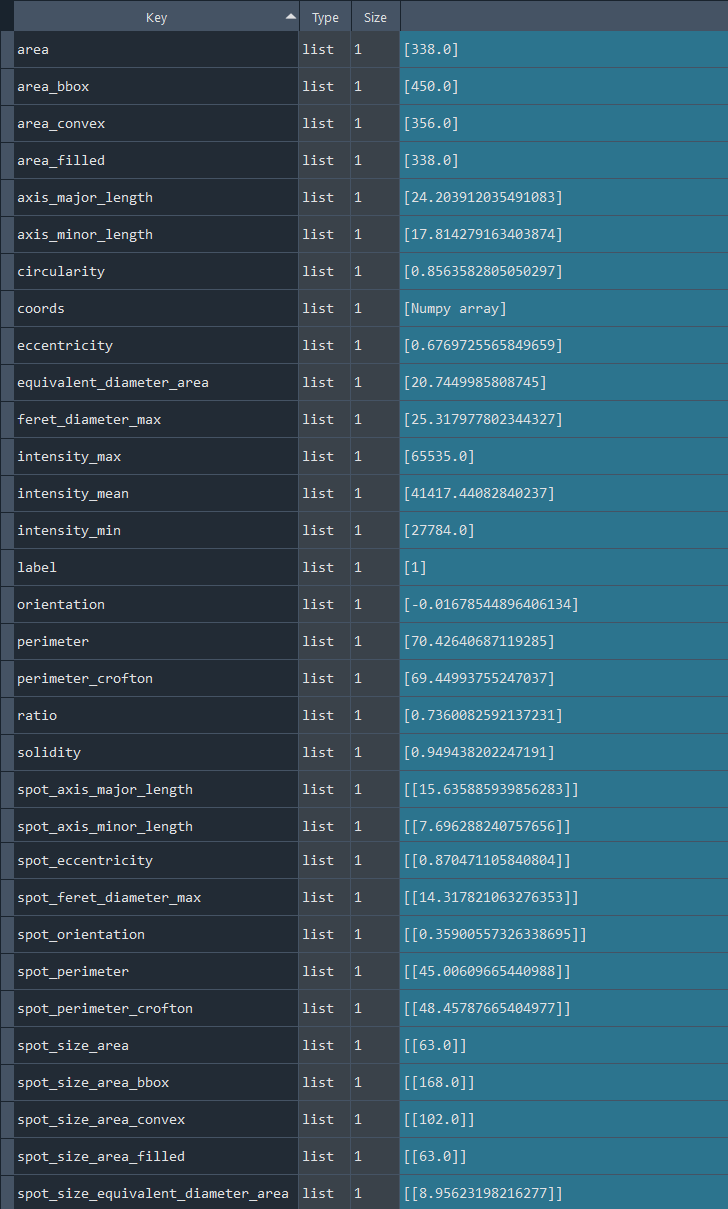
3.2.6 Analyzing nuclei chromatinization series
# If your parameters are correct for your data, you can run series analysis on more images
# Chromatinization CTRL CELLS
series_results_chromatinization = nf.series_analysis_chromatinization(path_to_images = 'test_data/flow_cytometry/ctrl',
file_extension = 'tif',
selected_id = [],
selection_opt = True,
include_img = False,
test_series = 0)
Dictionary with series nuclei chromatinization results:
# Chromatinization DISEASE CELLS
series_results_chromatinization2 = nf.series_analysis_chromatinization(path_to_images = 'test_data/flow_cytometry/dis',
file_extension = 'tif',
selected_id = [],
selection_opt = True,
include_img = False,
test_series = 0)
Dictionary with series nuclei chromatinization results:
3.2.7 Obtaining nuclei chromatinization series analysis results
from jimg_ncd.nuclei import NucleiDataManagement
# initiate class with NucleiFinder data
ndm = NucleiDataManagement(series_results_chromatinization, 'healthy_chromatinization')
ndm2 = NucleiDataManagement(series_results_chromatinization, 'disease_chromatinization')
# get data as data frame
df = ndm.get_data()
print(df)
ndm.save_results_df(path='')
df2 = ndm2.get_data()
print(df)
ndm2.save_results_df(path='')
Healthy results in data frame:
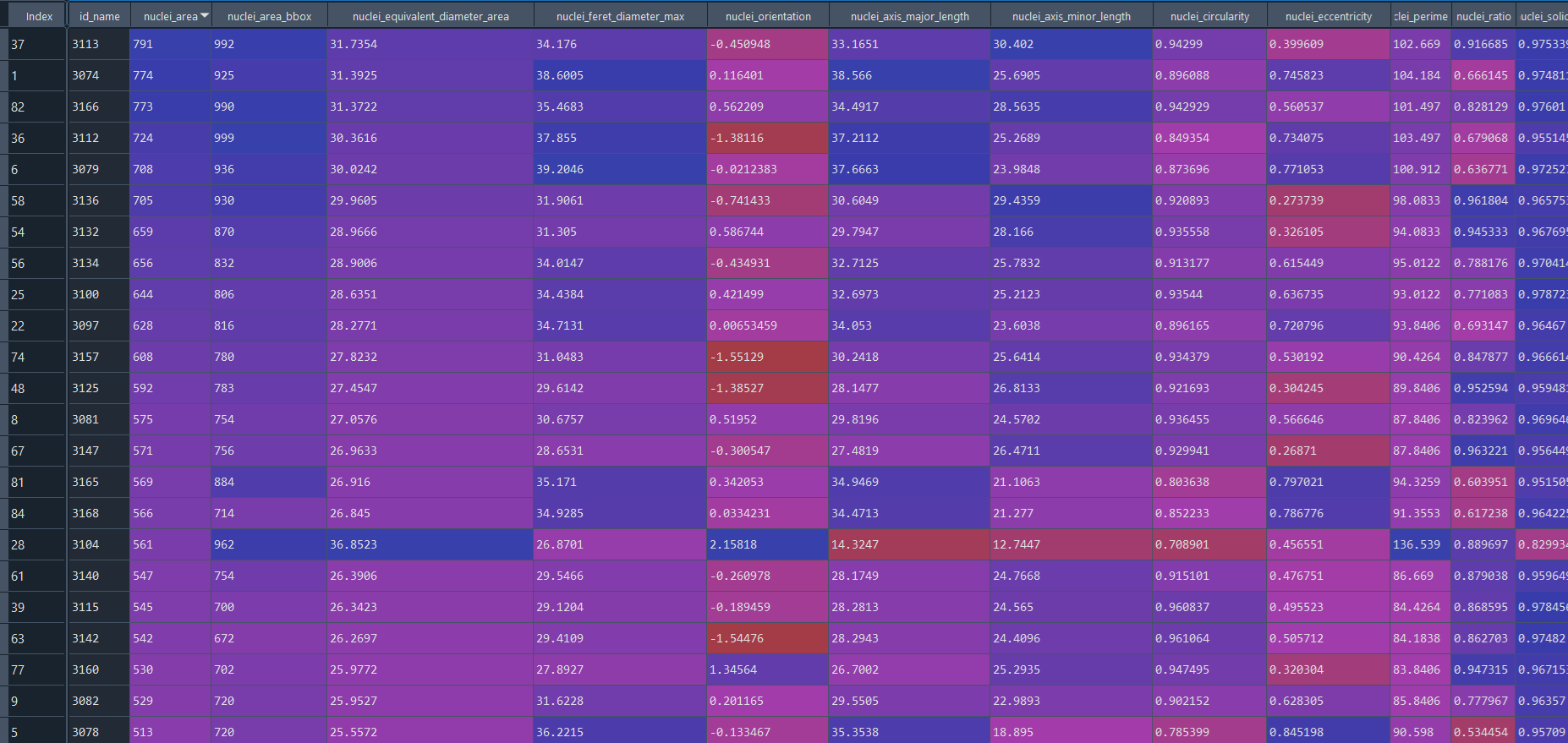
Disease results in data frame:
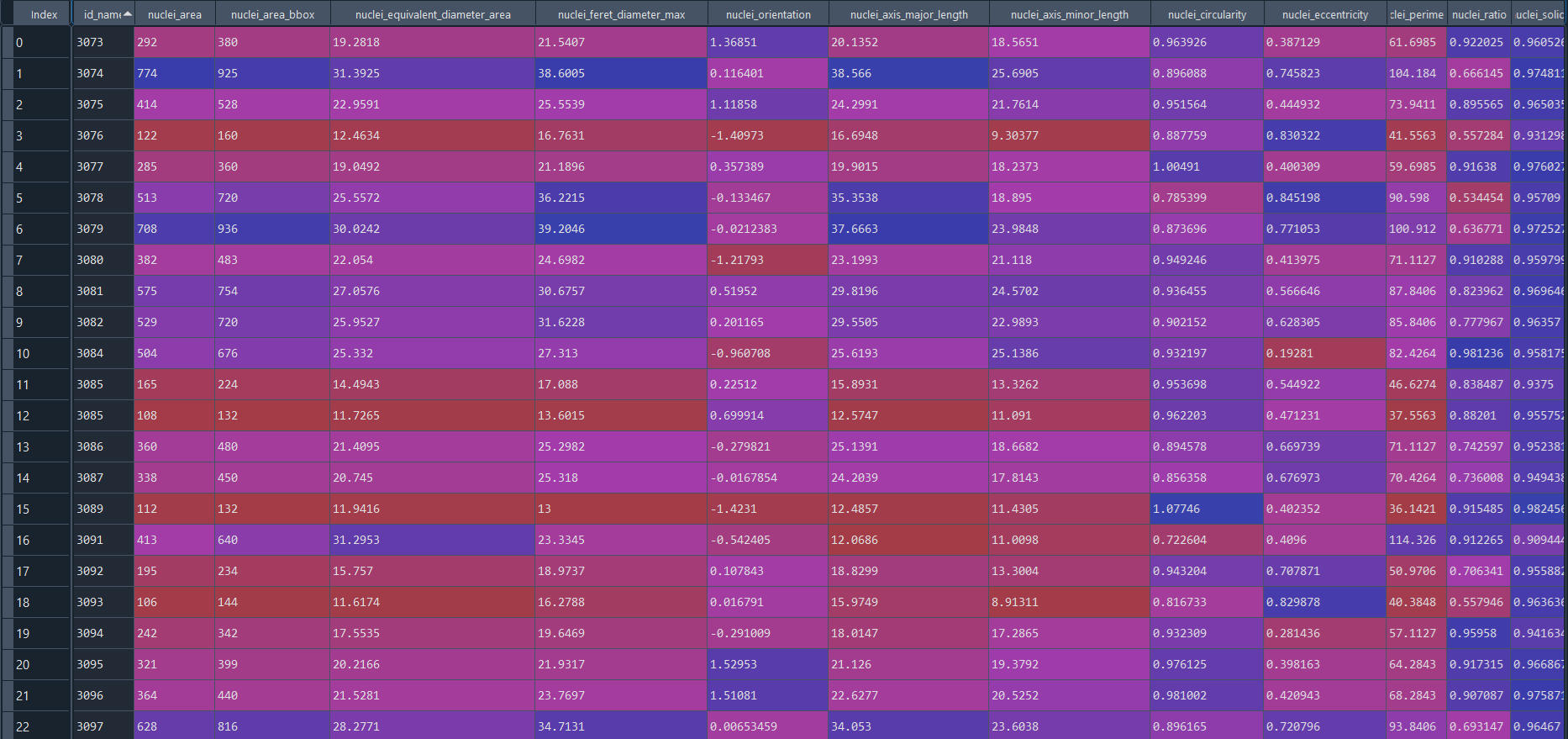
3.2.8 Concatenating nuclei chromatinization analysis with ImageStream (IS) data
# load IS data
import pandas as pd
healthy_data = pd.read_csv('test_data/flow_cytometry/ctrl.txt', sep='\t', header=1)
disease_data = pd.read_csv('test_data/flow_cytometry/dis.txt', sep='\t', header=1)
# select data with cell size info
selectes_columns = [
'Area_M01',
'Major Axis_M01',
'Minor Axis_M01',
'Aspect Ratio_M01',
'Diameter_M01',
'Area_M09',
'Major Axis_M09',
'Minor Axis_M09',
'Aspect Ratio_M09',
'Diameter_M09',
]
ndm.add_IS_data(healthy_data, IS_features=selectes_columns)
ndm2.add_IS_data(disease_data, IS_features=selectes_columns)
df_is = ndm.get_data_with_IS()
print(df_is)
ndm.save_results_df_with_IS(path = '')
df_is2 = ndm2.get_data_with_IS()
print(df_is2)
ndm2.save_results_df_with_IS(path = '')
# save projects
# NOTE:
# Image Stream (IS) data is NOT saved within the .nuc project file.
# After loading a project using `load_nuc_dict()`, IS data must be added again
# using the `add_IS_data()` method.
ndm.save_nuc_project(path='')
ndm2.save_nuc_project(path='')
Healthy results in data frame with IS data:
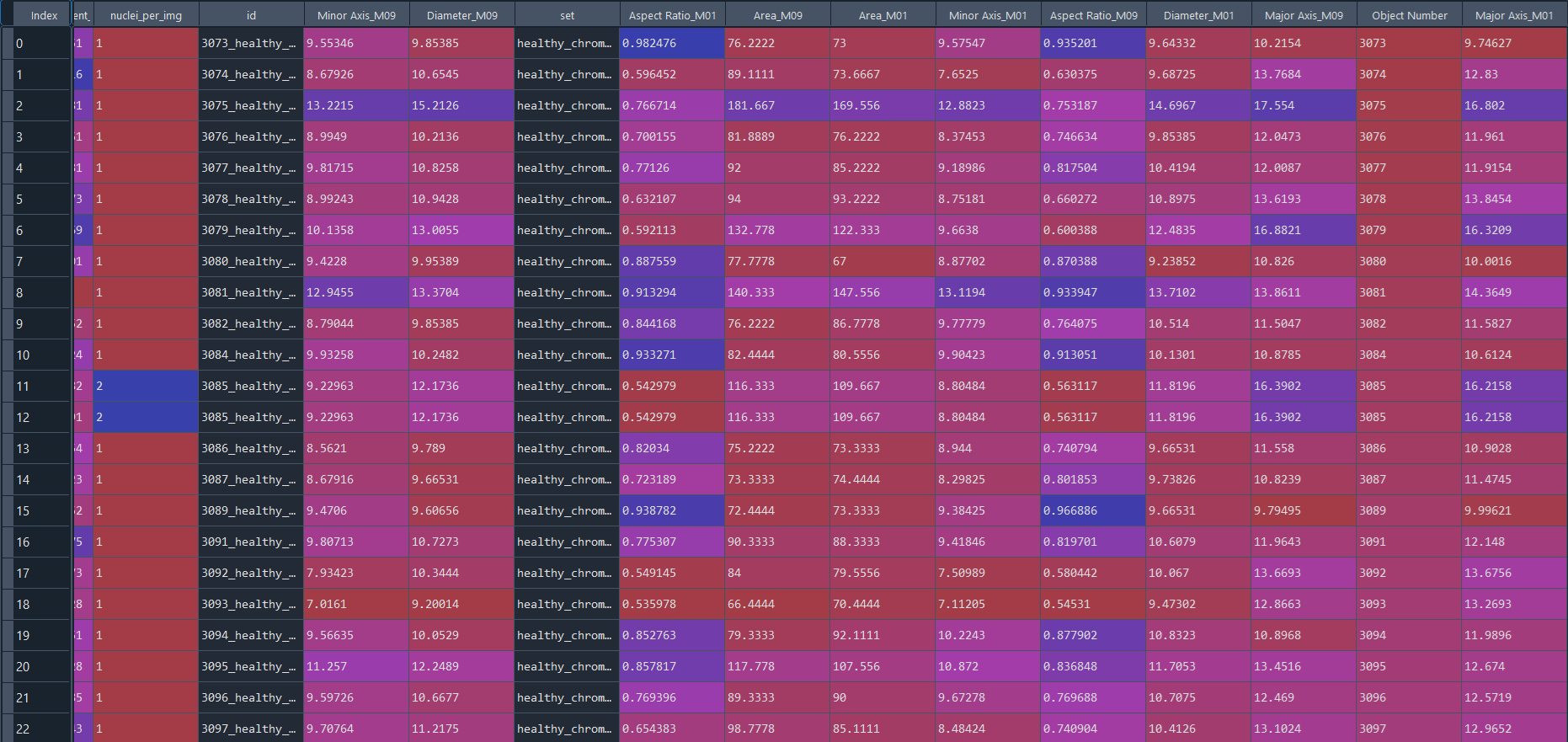
Disease results in data frame with IS data:
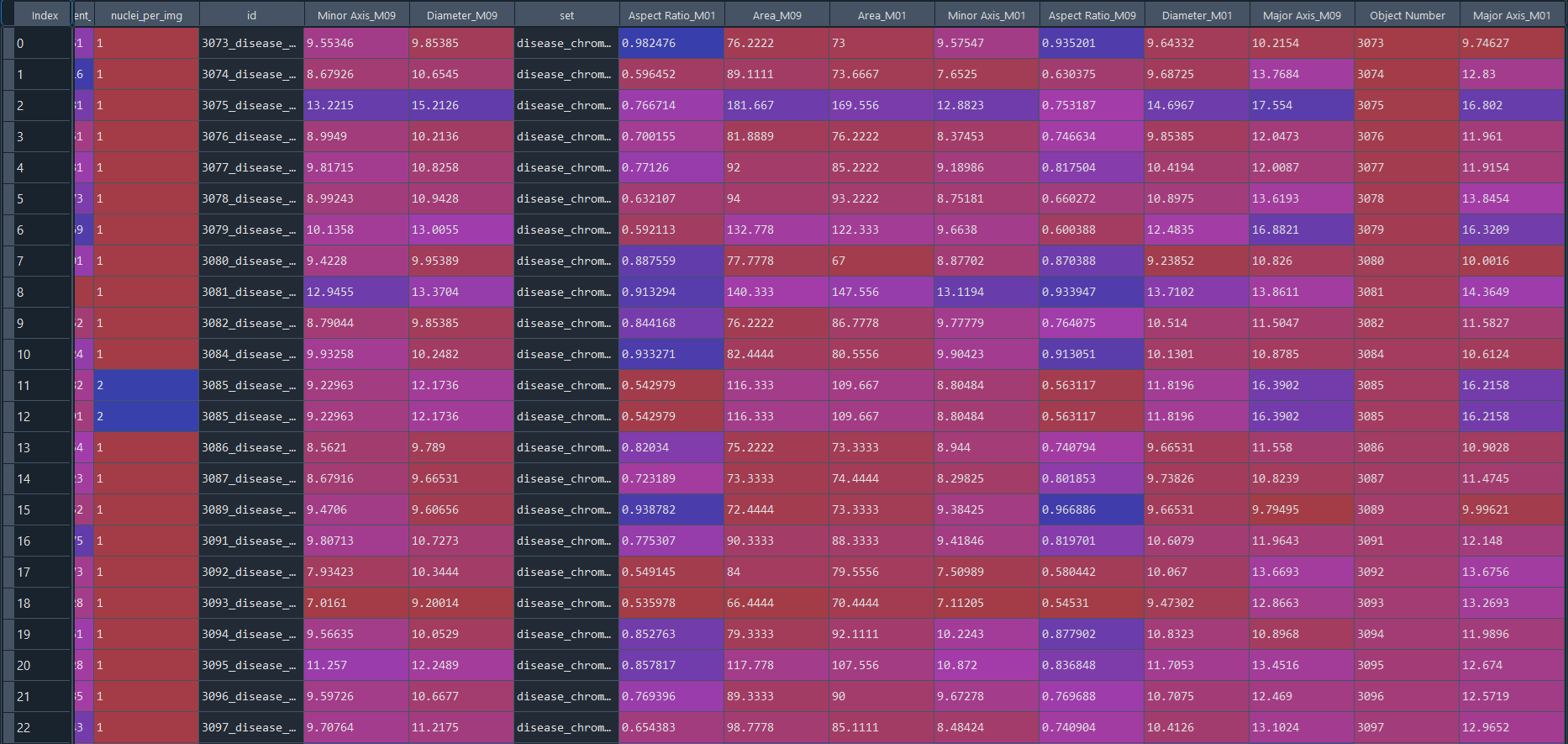
3.2.9 Combining projects from nuclei chromatinization analysi
from jimg_ncd.nuclei import NucleiDataManagement
# merge projects
ndm = NucleiDataManagement.load_nuc_dict('healthy_chromatinization.nuc')
ndm2 = NucleiDataManagement.load_nuc_dict('disease_chromatinization.nuc')
# load IS data
# NOTE:
# Image Stream (IS) data is NOT saved within the .nuc project file.
# After loading a project using `load_nuc_dict()`, IS data must be added again
# using the `add_IS_data()` method.
import pandas as pd
healthy_data = pd.read_csv('test_data/flow_cytometry/ctrl.txt', sep='\t', header=1)
disease_data = pd.read_csv('test_data/flow_cytometry/dis.txt', sep='\t', header=1)
# select data with cell size info
selectes_columns = [
'Area_M01',
'Major Axis_M01',
'Minor Axis_M01',
'Aspect Ratio_M01',
'Diameter_M01',
'Area_M09',
'Major Axis_M09',
'Minor Axis_M09',
'Aspect Ratio_M09',
'Diameter_M09',
]
ndm.add_IS_data(healthy_data, IS_features=selectes_columns)
ndm2.add_IS_data(disease_data, IS_features=selectes_columns)
ndm.add_experiment([ndm2])
df = ndm.get_mutual_experiments_data(inc_is=True)
print(df)
ndm.save_mutual_experiments(path='', inc_is=True)
Combined healthy and disease results in data frame with IS data:
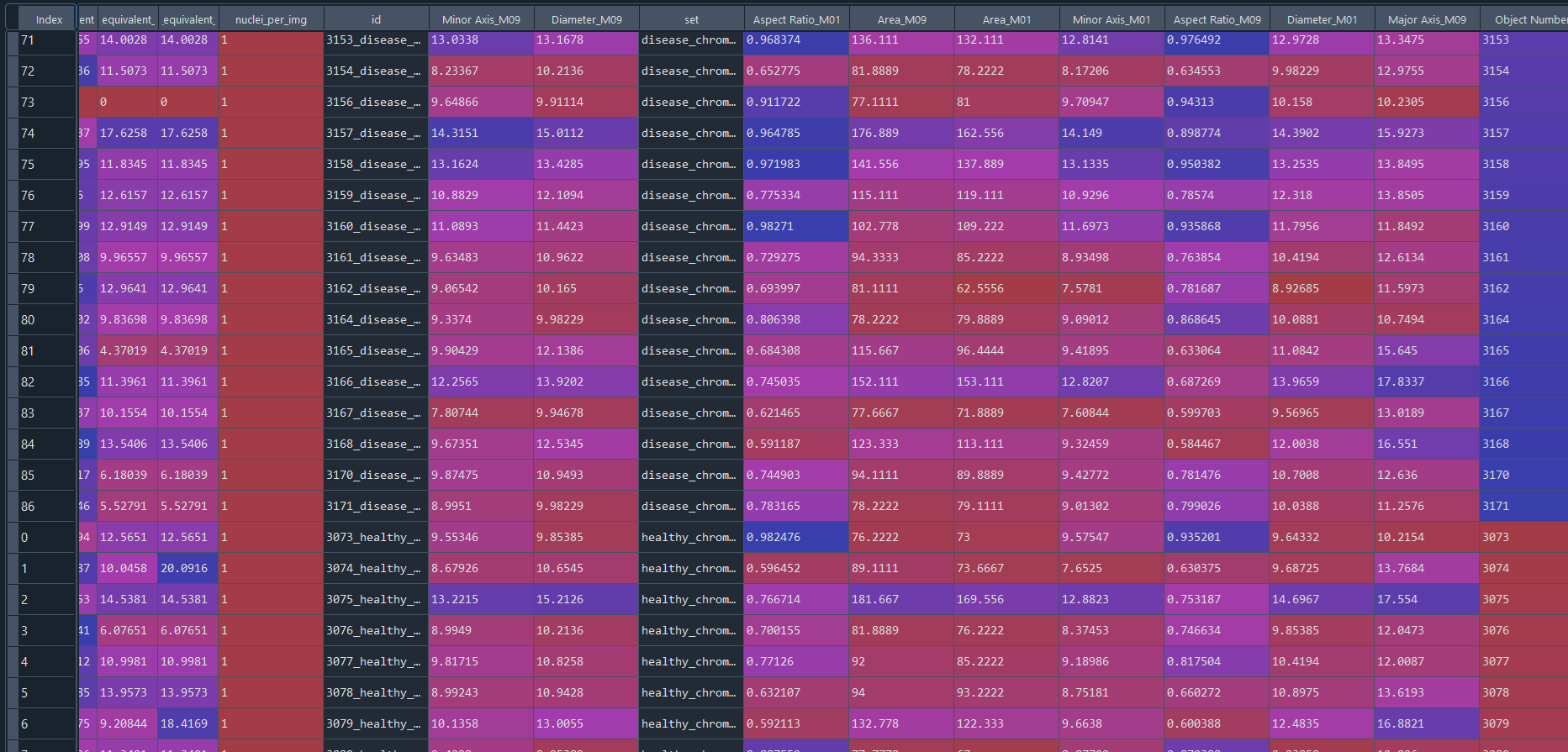
3.3 Clustering and DFA (Differential Feature Analysis) – Nuclei data
3.3.1 Selecting feature analysis (DFA) for separate experiments data
from jimg_ncd.nuclei import GroupAnalysis
import pandas as pd
data = pd.read_csv('healthy_chromatinization_disease_chromatinization_IS.csv', sep=',', header=0)
# initiate class
ga = GroupAnalysis.load_data(data, ids_col = 'id_name', set_col = 'set')
# check available groups for selection of differential features
ga.groups
# run DFA analysis on example sets
ga.DFA(meta_group_by = 'sets',
sets = {'disease':['disease_chromatinization'],
'ctrl':['healthy_chromatinization']},
n_proc = 5)
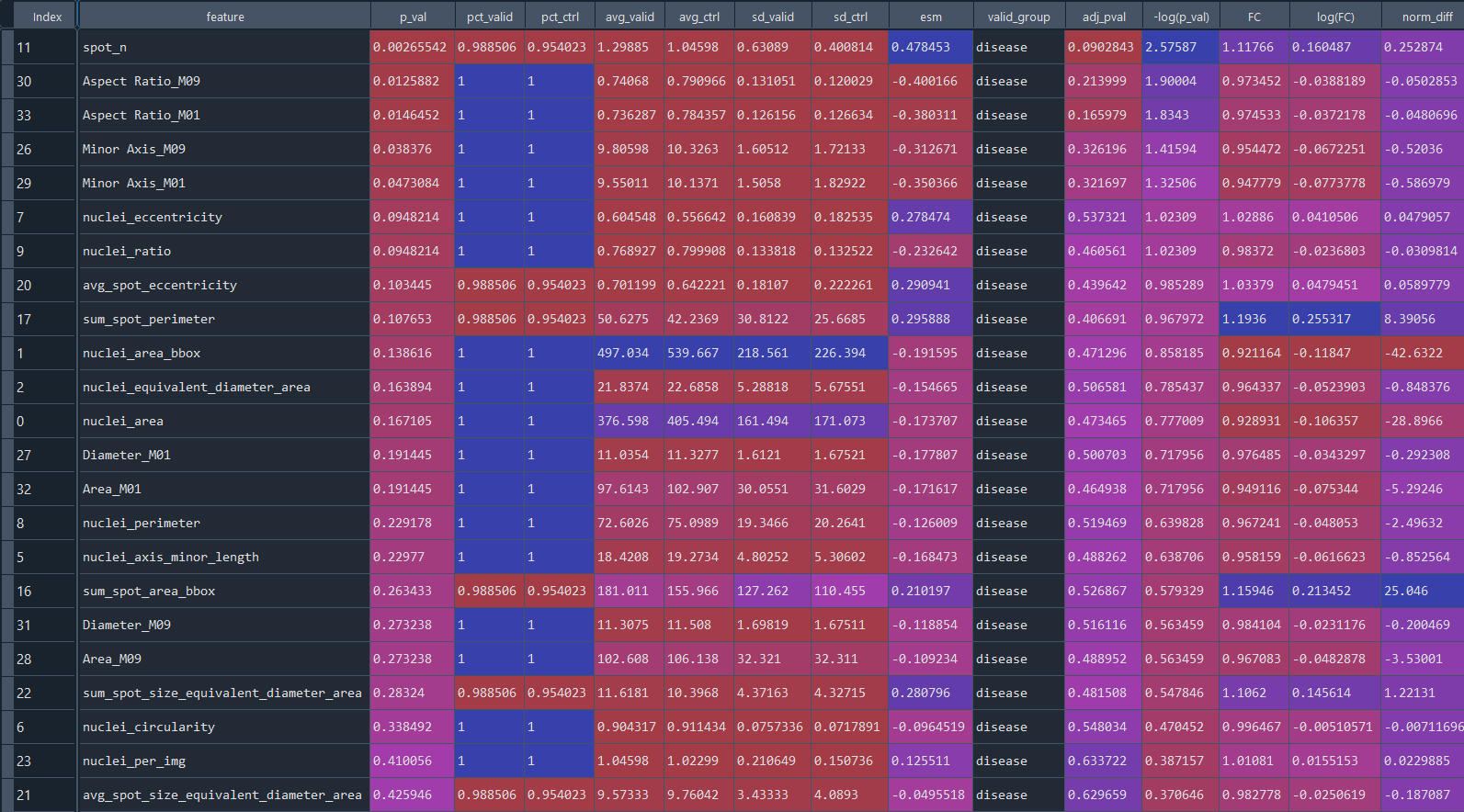
group_diff_features = ga.get_DFA()
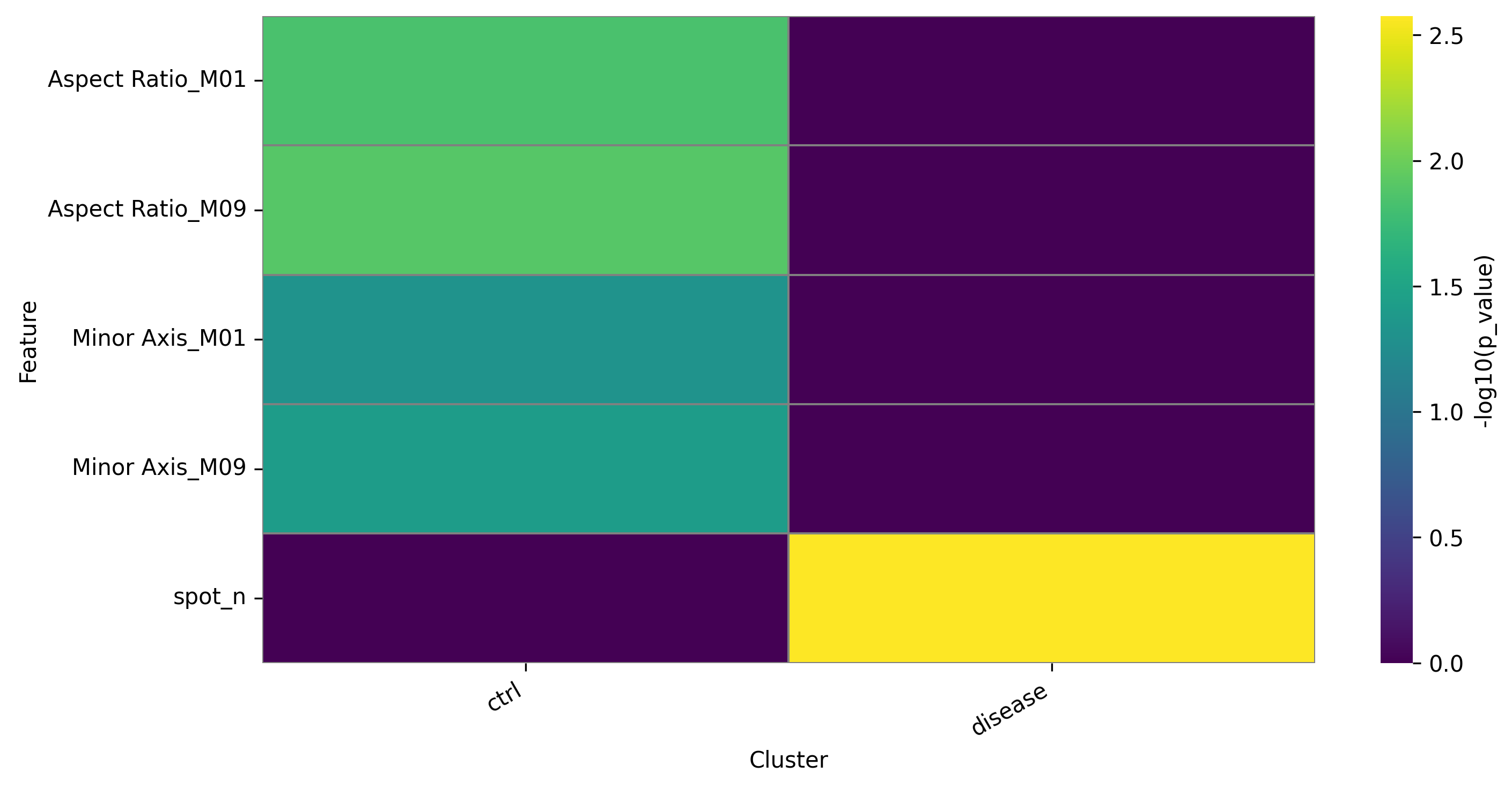
ga.heatmap_DFA()
fig = ga.get_DFA_plot()
fig.savefig('DFA_groups.png', dpi=300, bbox_inches='tight')
Data table presenting statistical analysis of differential features:
DFA heatmap for groups:
3.3.2 Filtering project data to selected features
# select differential features
diff_features = list(group_diff_features['feature'][group_diff_features['p_val'] <= 0.05])
ga.select_data(features_list = diff_features)
3.3.3 Performing data scaling and dimensionality reduction
# scale data
ga.data_scale()
# run PCA dimensionality reduction
ga.PCA()
# get PCA data, if required
pca_data = ga.get_PCA()
# run PC variance analysis
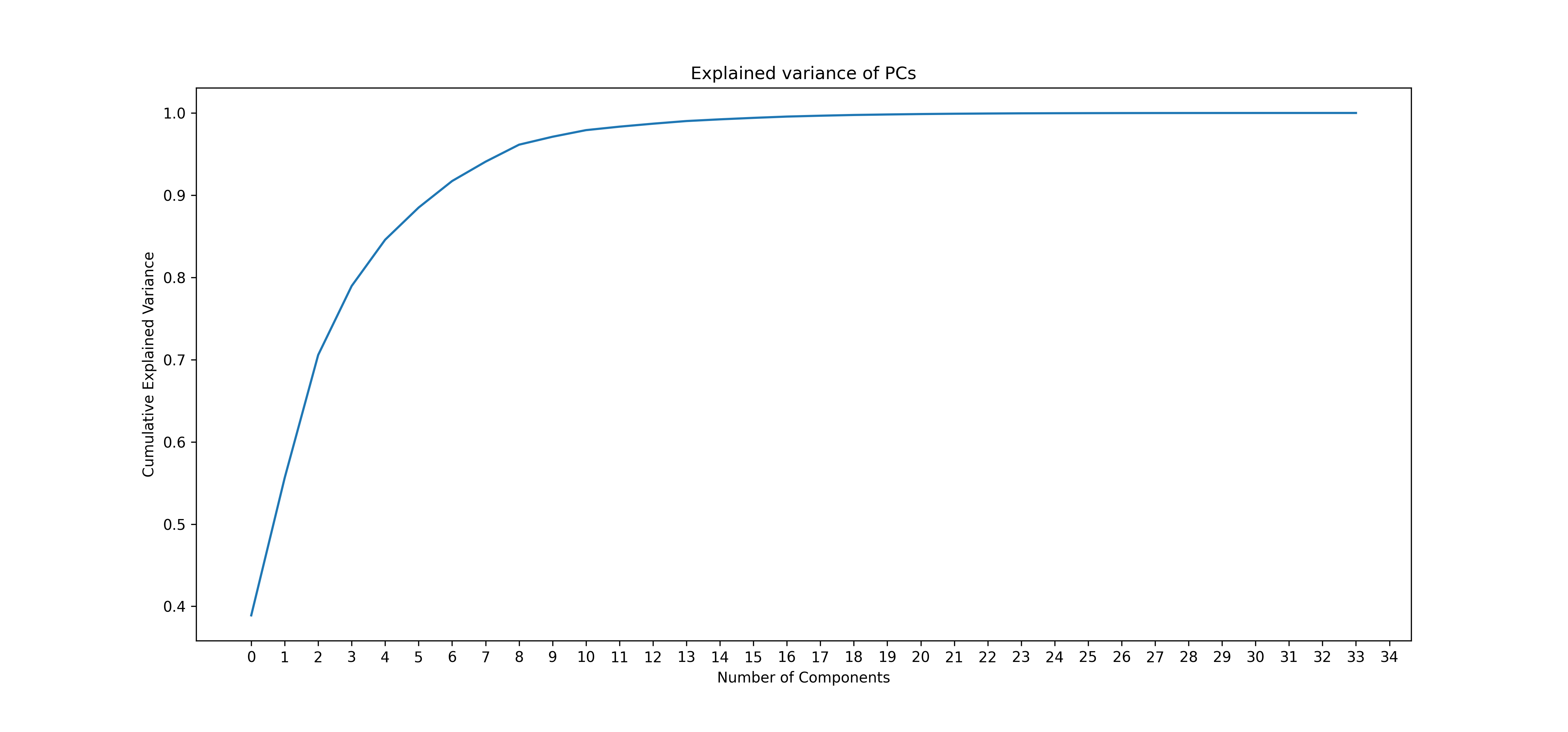
ga.var_plot()
# get var_data, if required
var_data = ga.get_var_data()
# get knee_plot, if required
knee_plot = ga.get_knee_plot(show = True)
knee_plot.savefig('knee_plot.png', dpi=300)
Knee plot - cumulative explanation of variance:
3.3.4 Performing UMAP & clustering
# run UMAP dimensionality reduction
ga.UMAP(PC_num = 8,
factorize_with_metadata = False,
harmonize_sets = True,
n_neighbors = 10,
min_dist = 0.01,
n_components = 2)
# get UMAP_data, if required
UMAP_data = ga.get_UMAP_data()
# get UMAP_plots, if required
UMAP_plots = ga.get_UMAP_plots(show = True)
UMAP_plots.keys()
UMAP_plots['PrimaryUMAP'].savefig('UMAP.png', dpi=300)
UMAP plot - sets:
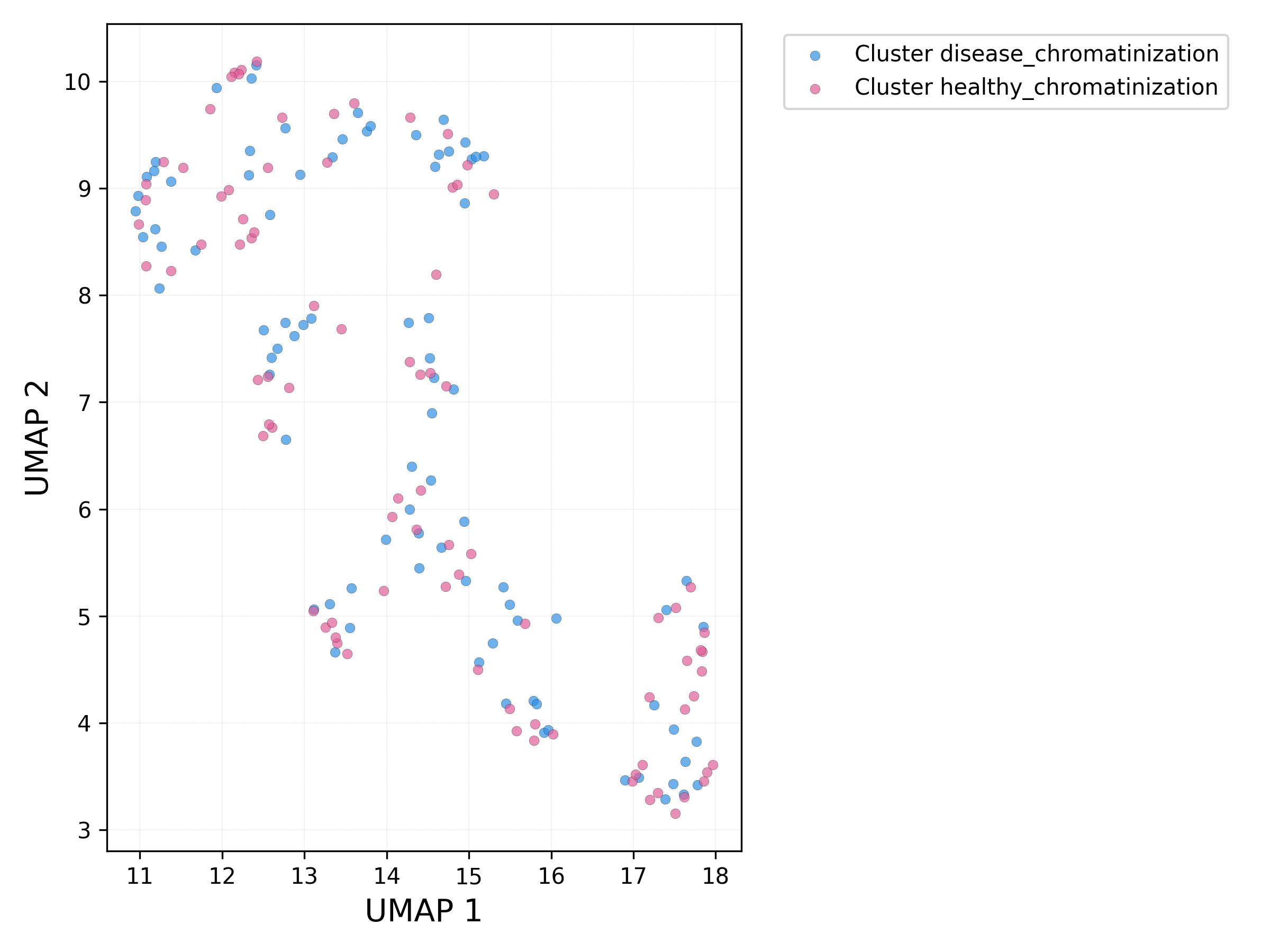
# run db_scan on UMAP components
ga.db_scan(eps = 0.5,
min_samples = 10)
# run UMAP_on_clusters
ga.UMAP_on_clusters(min_entities = 5)
# get UMAP_plots, if required
UMAP_plots = ga.get_UMAP_plots(show = True)
UMAP_plots.keys()
UMAP_plots['ClusterUMAP'].savefig('UMAP_clusters.png', dpi=300)
UMAP_plots['ClusterXSetsUMAP'].savefig('ClusterXSetsUMAP.png', dpi=300)
UMAP plot - db_scan clusters:
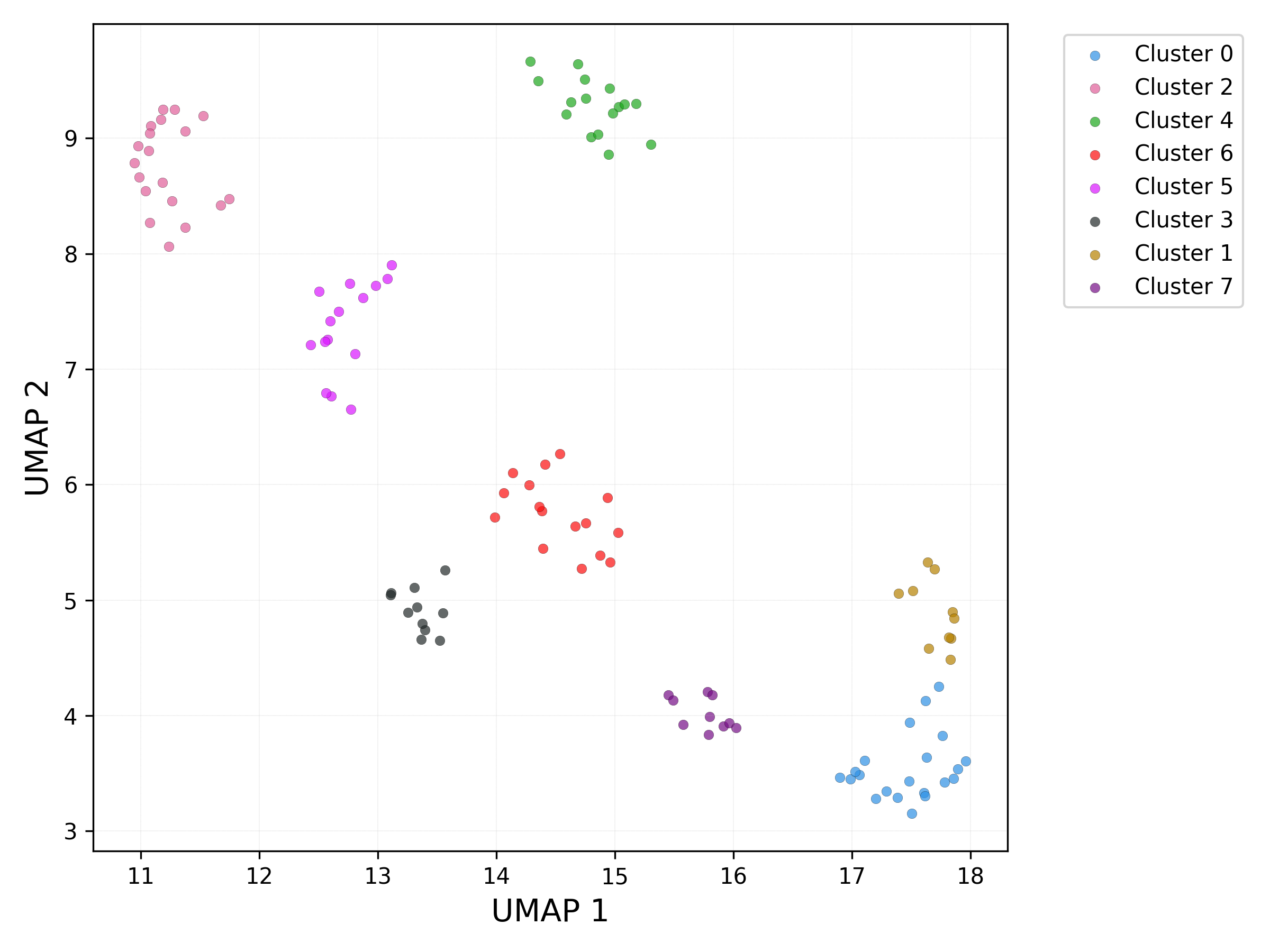
UMAP plot - set / cluster combination:
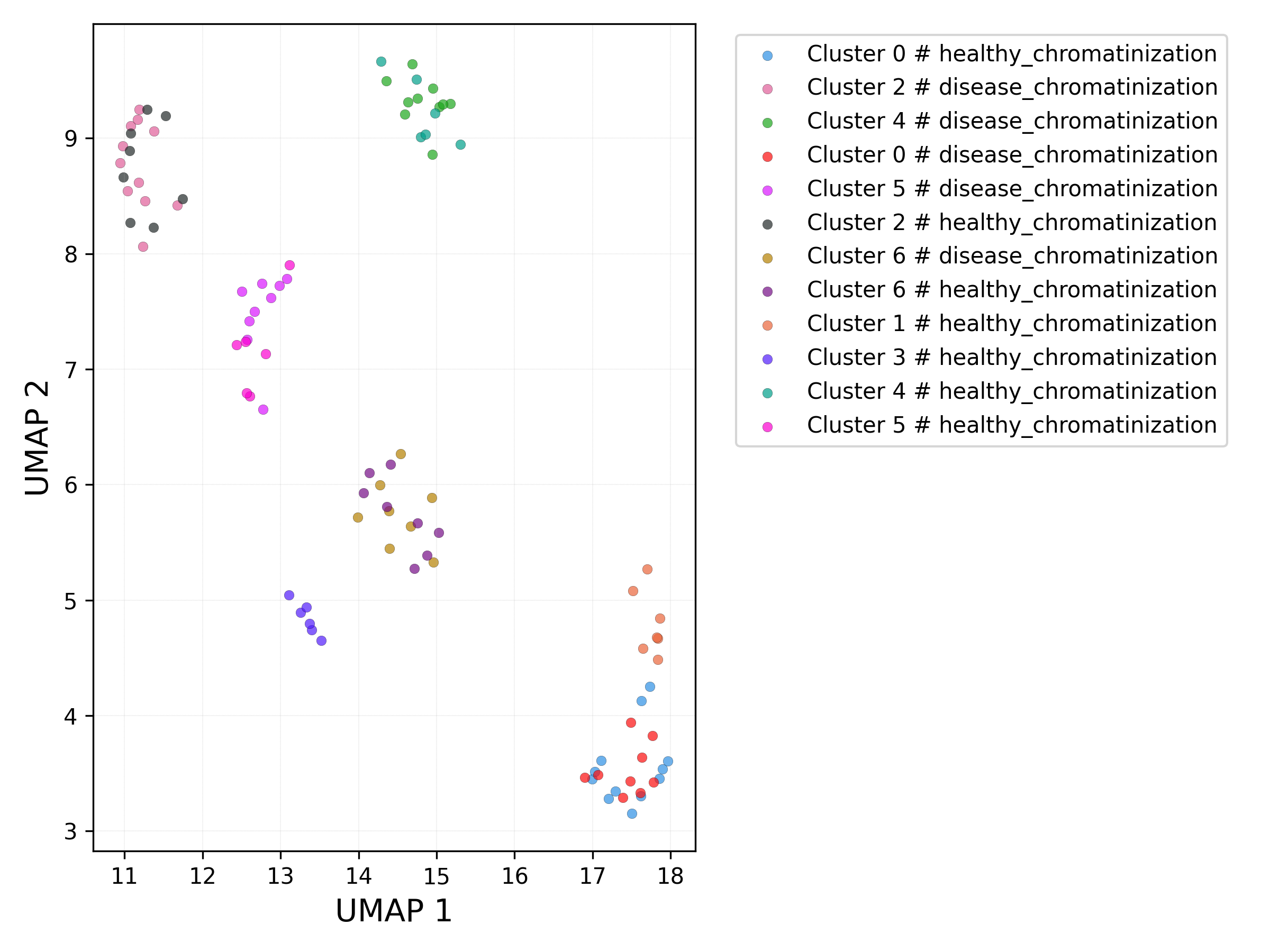
3.3.5 Obtaining complete data and metadata (clusters)
# get full_data [data + metadata], if required
full_data = ga.full_info()
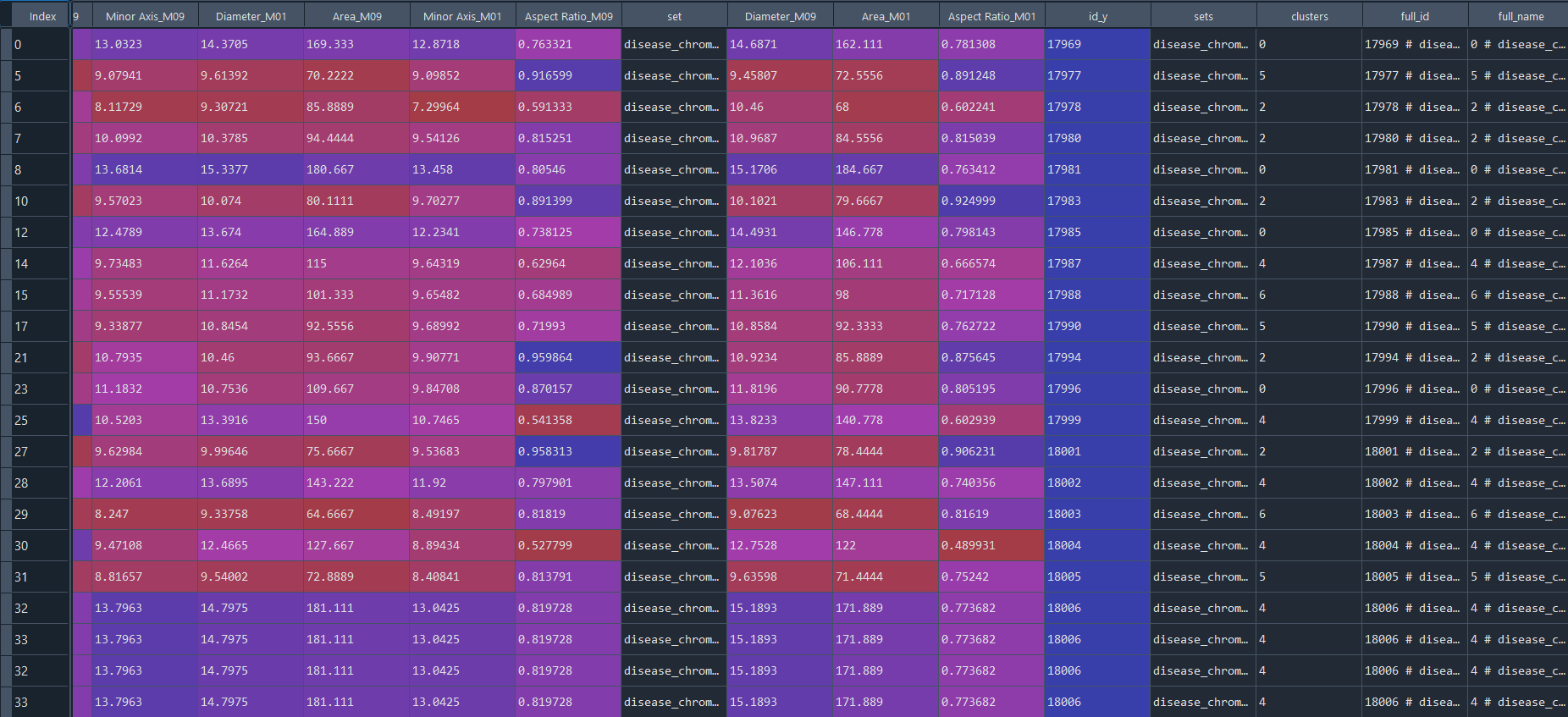
3.3.6 Preforming DFA analysis on clusters
# check available groups for selection of differential features
ga.groups
# run DFA analysis on finl clusters
ga.DFA(meta_group_by = 'full_name',
sets = {},
n_proc = 5)
dfa_clusters = ga.get_DFA()
ga.heatmap_DFA(top_n = 3)
fig = ga.get_DFA_plot()
fig.savefig('DFA_clusters.png', dpi=300, bbox_inches='tight')
Data table presenting statistical analysis of differential features for final clusters:
DFA heatmap for clusters:
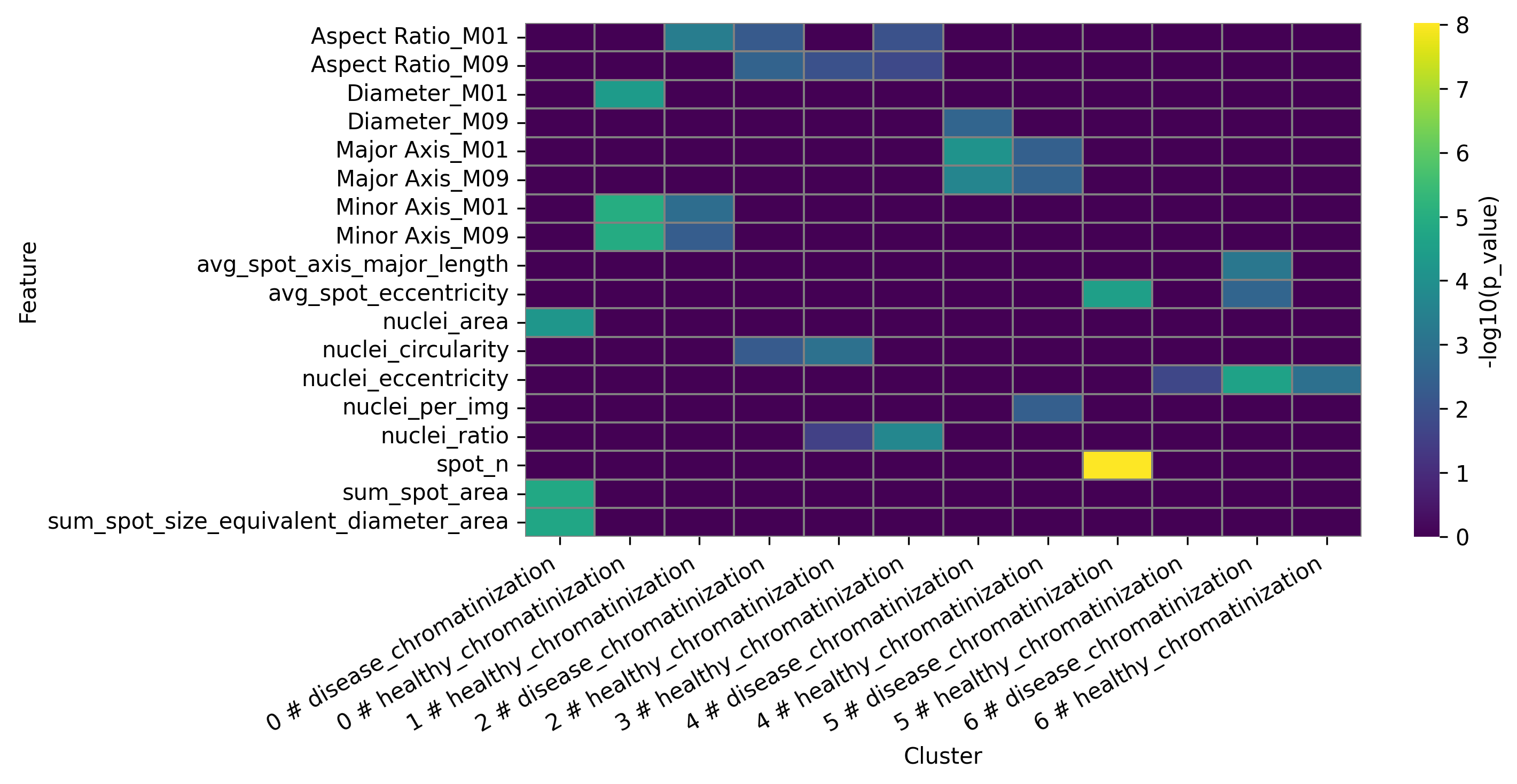
3.3.7 Preforming proportion analysis
ga.print_avaiable_features()
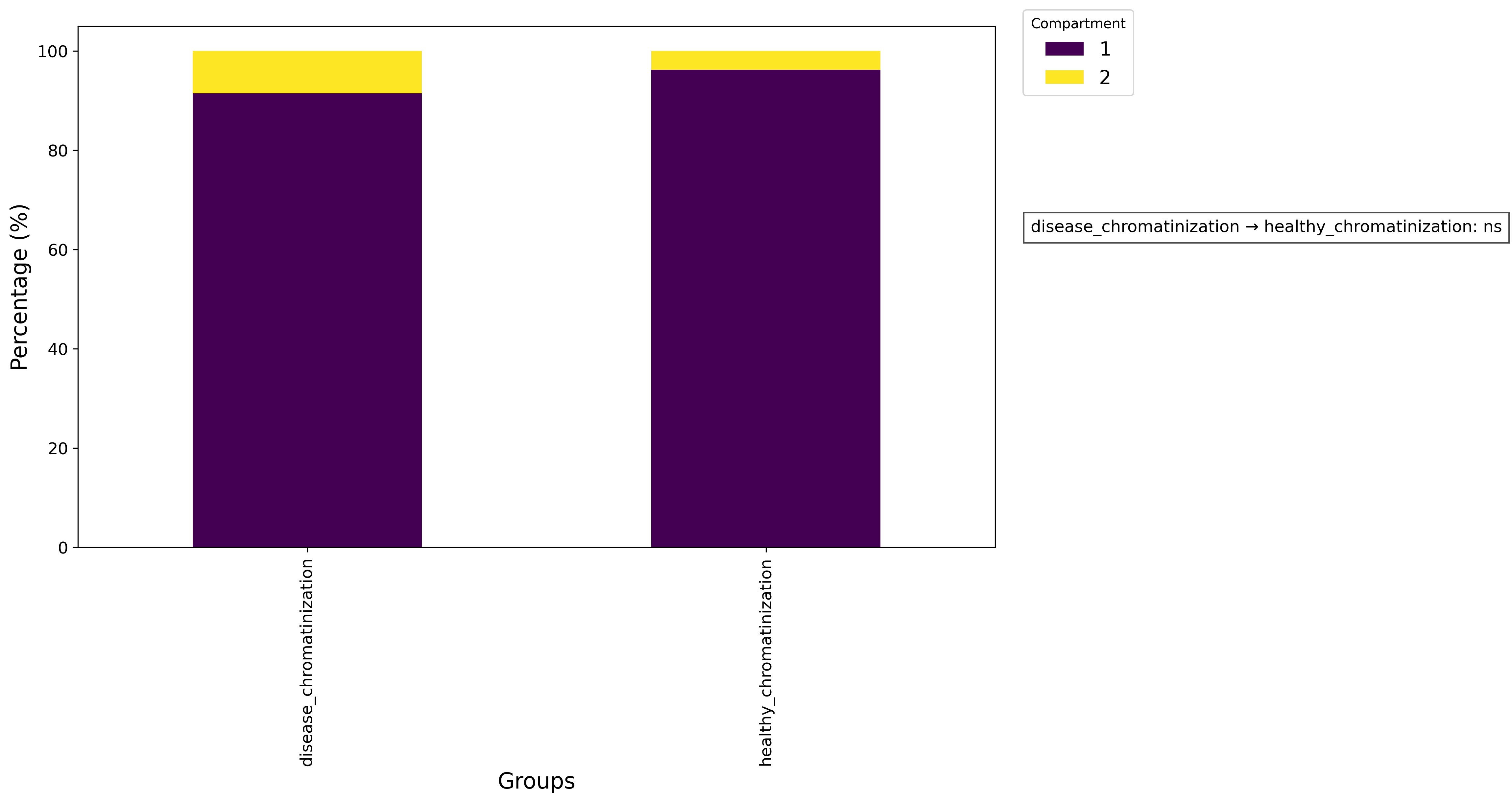
ga.proportion_analysis(grouping_col = 'sets',
val_col = 'nuclei_per_img',
grouping_dict = None,
omit = None)
pl = ga.get_proportion_plot(show = True)
pl.savefig('proportion.png', dpi=300, bbox_inches='tight')
ga.get_proportion_stats()
Nuclei per cell proportion plot:
3.4 ImagesManagement – nuclei data
3.4.1 Loading & saving raw data from project
from jimg_ncd.nuclei import ImagesManagement
# load dictionary data obtained from series_analysis_chromatinization()
# or series_analysis_nuclei() with include_img = True
indm = ImagesManagement.load_experimental_images(series_results_chromatinization, 'disease')
indm.save_raw(path_to_save = '')
3.4.2 Loading images IDs
from jimg_ncd.nuclei import ImagesManagement
# instead of loading dictionary data obtained from
# series_analysis_chromatinization() or series_analysis_nuclei()
# only image IDs to adjust can be loaded
images_ids = [1523, 1528, 1589, 1335]
indm = ImagesManagement.load_images_ids(images_ids = images_ids, experiment_name = 'disease')
3.4.3 Loading saved project with images
from jimg_ncd.nuclei import ImagesManagement
# loading whole project with data obtained from
# series_analysis_chromatinization() or series_analysis_nuclei()
# saved using self.save_raw(path_to_save = '')
indm = ImagesManagement.load_from_dict('disease.inuc.npz', 'disease')
3.4.4 Adjusting a series of images
indm.adjust_images(acronyme='bright',
color='gray',
path_to_images='test_data/flow_cytometry/dis',
fille_name_part='Ch9.ome',
eq = False,
clahe = False,
gamma = 0.7,
contrast = 1.2)
indm.adjust_images(acronyme='DAPI',
color='blue',
path_to_images='test_data/flow_cytometry/dis',
fille_name_part='Ch7.ome',
eq = True,
clahe = True,
gamma = 2,
contrast = 1.2)
3.4.5 Stitching images
# check available image acronyms defined in adjust_images
indm.get_included_acronyms()
# Stitch result images loaded from the project
# if they were loaded using:
# - 3.4.3 Loading a saved project with images
# - 3.4.1 Loading & saving raw data from the project
indm.image_stitching(acronyms=['bright'], to_results_image=True)
# you can also stitch different adjusted images with different acronyms,
# even if they were loaded using 3.4.2 Loading image IDs
indm.image_stitching(acronyms=['bright', 'DAPI'], to_results_image=False)
Stitched image:

3.4.6 Merging images
# check available image acronyms defined in adjust_images
indm.get_included_acronyms()
indm.image_merging(acronyms = ['bright', 'DAPI'],
ratio_list = [0.9,0.6])
Merged image:
3.4.7 Saving a series of images
# check available image acronyms defined in adjust_images, image_stitching and image_merging
indm.get_included_acronyms()
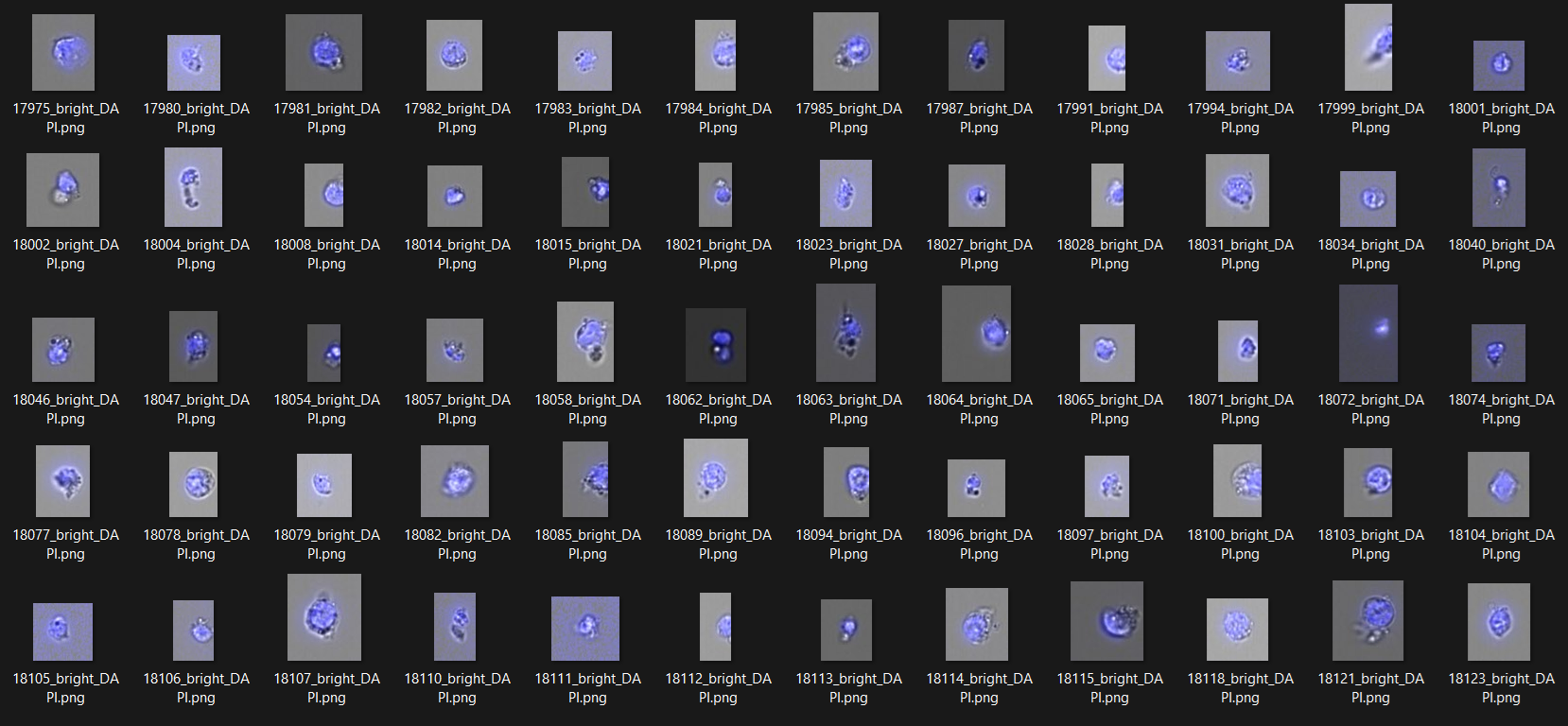
indm.save_prepared_images(acronyme = 'stitched_bright', path_to_save = '')
indm.save_prepared_images(acronyme = 'stitched_bright_DAPI', path_to_save = '')
indm.save_prepared_images(acronyme = 'merged_bright_DAPI', path_to_save = '')
Saved images:

Have fun JBS
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file jimg_ncd-0.1.10.tar.gz.
File metadata
- Download URL: jimg_ncd-0.1.10.tar.gz
- Upload date:
- Size: 53.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e1be8c17c7a2d048fe7e5884293bc38059a807ff690c50420dfb8ff160069999
|
|
| MD5 |
56516f51c75fecd6cb10cea278f15060
|
|
| BLAKE2b-256 |
e3c4d2f3e16147f4f950a448159ecbd6637b11e6ab6e07d214e3dc9f679a3ab8
|
File details
Details for the file jimg_ncd-0.1.10-py3-none-any.whl.
File metadata
- Download URL: jimg_ncd-0.1.10-py3-none-any.whl
- Upload date:
- Size: 51.0 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
14ac8d6df875e4575d2eab6b9a0d830dd3759ca59c4e56d9291c1564a6506824
|
|
| MD5 |
198f7233a3765530696a18dd50b96b48
|
|
| BLAKE2b-256 |
4a4ab16f067663ccc125b6bad37c6ebd7bad2c3d5d781d77a8170652bca82a78
|







