A collection of scripts designed to process Kraken2 reports and convert them into CSV format.
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
KrakenParser: Convert Kraken2 Reports to CSV



Overview
KrakenParser is a collection of scripts designed to process Kraken2 reports and convert them into CSV format. This pipeline extracts taxonomic abundance data at six levels:
- Phylum
- Class
- Order
- Family
- Genus
- Species
Installation
# Linux / WSL / macOS
conda create -n krakenparser pip -y
conda activate krakenparser
pip install krakenparser
Usage Guide
Full Pipeline
KrakenParser -i data/kreports -o results/
This will:
- Convert Kraken2 reports to MPA format
- Combine MPA files into a single file
- Extract taxonomic levels into separate text files
- Process extracted text files
- Convert them into CSV format
- Calculate relative abundance
- Calculate α & β-diversities
[!TIP] After the pipeline finishes, the output window will remind you about calibrating rarefaction depth for β-diversity and re-running relative abundance normalization before visualization — with ready-to-paste example commands tailored to your output paths.
Full help output
Usage: KrakenParser [OPTIONS] COMMAND [ARGS]...
KrakenParser: Convert Kraken2 Reports to CSV and analyze microbial diversity.
To execute the full pipeline automatically, just use the global options.
Alternatively, you can run specific parts of the pipeline manually in the
following order:
mpa ➔ combine ➔ split ➔ process ➔ csv ➔ relabund ➔ diversity
Each step behaves as an independent tool. Type 'krakenparser <command> --help'
to see options for a specific step.
╭─ Options ────────────────────────────────────────────────────────────────────╮
│ --input -i PATH Directory containing Kraken2 report │
│ files. │
│ --output -o PATH Output directory. │
│ --viruses -viruses Extract only VIRUSES domain taxa in │
│ the pipeline. │
│ --bacteria -bacteria Extract only BACTERIA domain taxa in │
│ the pipeline. │
│ --fungi -fungi Extract only FUNGI kingdom taxa in │
│ the pipeline. │
│ --archaea -archaea Extract only ARCHAEA domain taxa in │
│ the pipeline. │
│ --keep-human -keep-human Do not filter human-related taxa. │
│ --version -V Show version and exit. │
│ --depth -d INTEGER Rarefaction depth for β-diversity. │
│ [default: 1000] │
│ --seed -s INTEGER Random seed for reproducible │
│ rarefaction. │
│ --overwrite -overwrite Overwrite the output directory if it │
│ already exists. │
│ --help -h Show this message and exit. │
╰──────────────────────────────────────────────────────────────────────────────╯
╭─ Advanced (Step-by-step pipeline control) ───────────────────────────────────╮
│ mpa Convert a Kraken2 report to MetaPhlAn (MPA) format. │
│ combine Combine MPA files into a single tab-delimited table. │
│ split Split a combined MPA table into per-rank TXT files. │
│ process Reads a source file, processes its first line, modifies taxa │
│ names in a destination file, and updates it. │
│ csv Reads a TXT file, reorganizes the data, and converts it into a │
│ CSV file. │
│ relabund Calculates taxa relative abundance and saves it to a CSV file. │
│ diversity Calculate α & β-diversities for microbial communities. │
╰──────────────────────────────────────────────────────────────────────────────╯
🔗 Please visit KrakenParser wiki page
Advanced step-by-step mode
Advanced usage
Each step behaves as an independent tool. Type krakenparser <command> --help to see options for a specific step.
Step 1: Convert Kraken2 Reports to MPA Format
# Batch mode (directory)
KrakenParser mpa -i data/kreports -o data/intermediate/mpa
# Single file
KrakenParser mpa -r data/kreports/sample.kreport -o data/intermediate/mpa/sample.MPA.TXT
Converts Kraken2 .kreport files into MPA format.
Step 2: Combine MPA Files
KrakenParser combine -i data/intermediate/mpa/* -o data/intermediate/COMBINED.txt
Merges multiple MPA files into a single combined table.
Step 3: Extract Taxonomic Levels
KrakenParser split -i data/intermediate/COMBINED.txt -o data/intermediate
By default, human-related taxa (Homo sapiens, Hominidae, Primates, Mammalia, Chordata) are removed. To keep them:
KrakenParser split -i data/intermediate/COMBINED.txt -o data/intermediate --keep-human
To inspect the Viruses domain only:
KrakenParser split -i data/intermediate/COMBINED.txt -o data/counts_viruses --viruses-only
Same for Bacteria and Archaea domains and Fungi kingdom (--bacteria-only; --archaea-only & --fungi-only)
Step 4: Process Extracted Taxonomic Data
KrakenParser process -i data/intermediate/COMBINED.txt -o data/intermediate/txt/counts_phylum.txt
Repeat on other 5 taxonomical levels (class, order, family, genus, species) or wrap process in a loop.
Cleans up taxonomic names: removes prefixes (s__, g__, etc.) and replaces underscores with spaces.
Step 5: Convert TXT to CSV
KrakenParser csv -i data/intermediate/txt/counts_phylum.txt -o data/counts/counts_phylum.csv
Repeat on other 5 taxonomical levels or wrap in a loop. Transposes data so that sample names become rows.
Step 6: Calculate Relative Abundance
KrakenParser relabund -i data/counts/counts_phylum.csv -o data/rel_abund/ra_phylum.csv
Repeat on other 5 taxonomical levels or wrap in a loop.
With "Other" grouping:
KrakenParser relabund -i data/counts/counts_phylum.csv -o data/rel_abund/ra_phylum.csv -O 3.5
Groups all taxa with abundance < 3.5 % into Other (<3.5%).
Step 7: Calculate α & β-Diversities
KrakenParser diversity -i data/counts/counts_species.csv -o data/diversity
With a custom rarefaction depth:
KrakenParser diversity -i data/counts/counts_species.csv -o data/diversity -d 750
For reproducible results (fix the seed to get the same matrix every run):
KrakenParser diversity -i data/counts/counts_species.csv -o data/diversity -s 42
Output example
Total abundance output
counts_phylum.csv parsed from 9 kraken2 reports of metagenomic samples using KrakenParser:
Sample_id,Calditrichota,Caldisericota,Thermosulfidibacterota,Elusimicrobiota,Candidatus Fervidibacterota,Lentisphaerota,Kiritimatiellota,Vulcanimicrobiota,Thermodesulfobiota,Atribacterota,Dictyoglomota,Nitrospinota,Chrysiogenota,Coprothermobacterota,Aquificota,Thermotogota,Bdellovibrionota,Nitrospirota,Deferribacterota,Synergistota,Myxococcota,Acidobacteriota,Candidatus Bipolaricaulota,Candidatus Saccharibacteria,Candidatus Absconditabacteria,Fusobacteriota,Spirochaetota,Candidatus Omnitrophota,Chlamydiota,Verrucomicrobiota,Planctomycetota,Thermodesulfobacteriota,Campylobacterota,Candidatus Cloacimonadota,Fibrobacterota,Gemmatimonadota,Balneolota,Rhodothermota,Ignavibacteriota,Chlorobiota,Bacteroidota,Deinococcota,Thermomicrobiota,Armatimonadota,Chloroflexota,Cyanobacteriota,Mycoplasmatota,Actinomycetota,Bacillota,Pseudomonadota,Heterolobosea,Parabasalia,Fornicata,Evosea,Bacillariophyta,Cercozoa,Euglenozoa,Apicomplexa,Microsporidia,Basidiomycota,Ascomycota,Nanoarchaeota,Candidatus Micrarchaeota,Candidatus Thermoplasmatota,Candidatus Lokiarchaeota,Nitrososphaerota,Euryarchaeota,Thermoproteota,Hofneiviricota,Artverviricota,Nucleocytoviricota,Cossaviricota,Kitrinoviricota,Negarnaviricota,Lenarviricota,Pisuviricota,Peploviricota,Uroviricota
X1,0,0,0,0,0,0,0,0,1,1,1,1,2,3,4,5,7,8,9,17,23,25,5,13,22,47,54,1,6,27,31,128,151,2,6,13,1,3,7,44,14991,7,9,11,61,414,449,3551,55304,438645,0,0,0,0,0,0,1,22,0,4,15,0,0,0,0,0,3,191,0,0,1,88,0,0,0,161,0,1241
X2,1,4,14,20,5,12,15,6,8,15,2,15,109,68,182,97,79,196,70,272,331,149,36,77,35,562,1237,21,33,129,427,1044,543,8,98,25,16,45,11,1043,41374,160,28,161,1348,1196,2709,15864,431170,2747842,22,7,301,373,134,136,107,3239,54,1151,2905,0,0,3,5,6,7,410,0,0,0,736,0,3,11,26,1,1552
...
X8,1,19,0,47,0,1,6,20,28,0,1,1,47,7,336,110,30,32,10,93,85,48,9,7,7,154,386,0,14,19,106,358,242,14,5,134,15,11,7,18,54057,106,10,24,212,340,1128,16220,567908,650264,95,4,193,402,314,300,187,4376,37,9796,8653,0,1,0,1,5,23,1778,1,1,0,1,1,4,66,30,4,1263
X9,0,3,2,16,7,1,23,12,10,9,1,2,134,40,390,289,29,372,27,81,150,90,9,88,32,287,881,14,33,60,319,1045,328,15,22,22,10,72,8,63,35301,127,15,48,412,935,2343,11500,380765,2613854,0,0,0,0,0,0,5,74,0,38,40,3,0,0,0,1,3,275,0,0,0,0,0,2,118,25,0,1675
Relative abundance output
ra_phylum.csv calculated from 9 kraken2 reports of metagenomic samples using KrakenParser:
Sample_id,taxon,rel_abund_perc
X1,Pseudomonadota,85.03558294577552
X1,Bacillota,10.72121619814011
X1,Other (<4.0%),4.243200856084384
X2,Pseudomonadota,84.28702055549813
X2,Bacillota,13.225663867469137
X2,Other (<4.0%),2.487315577032736
...
X8,Pseudomonadota,49.25373021277305
X8,Bacillota,43.01574040339849
X8,Bacteroidota,4.094504530639667
X8,Other (<4.0%),3.6360248531887933
X9,Pseudomonadota,85.62839981589192
X9,Bacillota,12.473649123439218
X9,Other (<4.0%),1.8979510606688494
α-diversity output
alpha_div.csv calculated from 9 kraken2 reports of metagenomic samples using KrakenParser:
Sample,Shannon,Pielou,Chao1
X1,3.911345447107001,0.5269245043289149,2274.533185840708
X2,3.9944130792536563,0.4906424221265042,4155.0
...
X8,3.442077115880119,0.42753293021330063,4177.251358695652
X9,4.033664950188261,0.5050385978575492,3492.16
β-diversity output
beta_div_bray.csv calculated from 9 kraken2 reports of metagenomic samples using KrakenParser:
,X1,X2,...,X8,X9
X1,0.0,0.398,...,0.61,0.353
X2,0.398,0.0,...,0.723,0.388
...
X8,0.61,0.723,...,0.0,0.665
X9,0.353,0.388,...,0.665,0.0
beta_div_jaccard.csv calculated from 9 kraken2 reports of metagenomic samples using KrakenParser:
,X1,X2,...,X8,X9
X1,0.0,0.7073170731707317,...,0.8223938223938224,0.7232472324723247
X2,0.7073170731707317,0.0,...,0.835016835016835,0.7352941176470589
...
X8,0.8223938223938224,0.835016835016835,...,0.0,0.8066914498141264
X9,0.7232472324723247,0.7352941176470589,...,0.8066914498141264,0.0
Visualization examples gallery
| Stacked Barplot | Streamgraph |
|---|---|
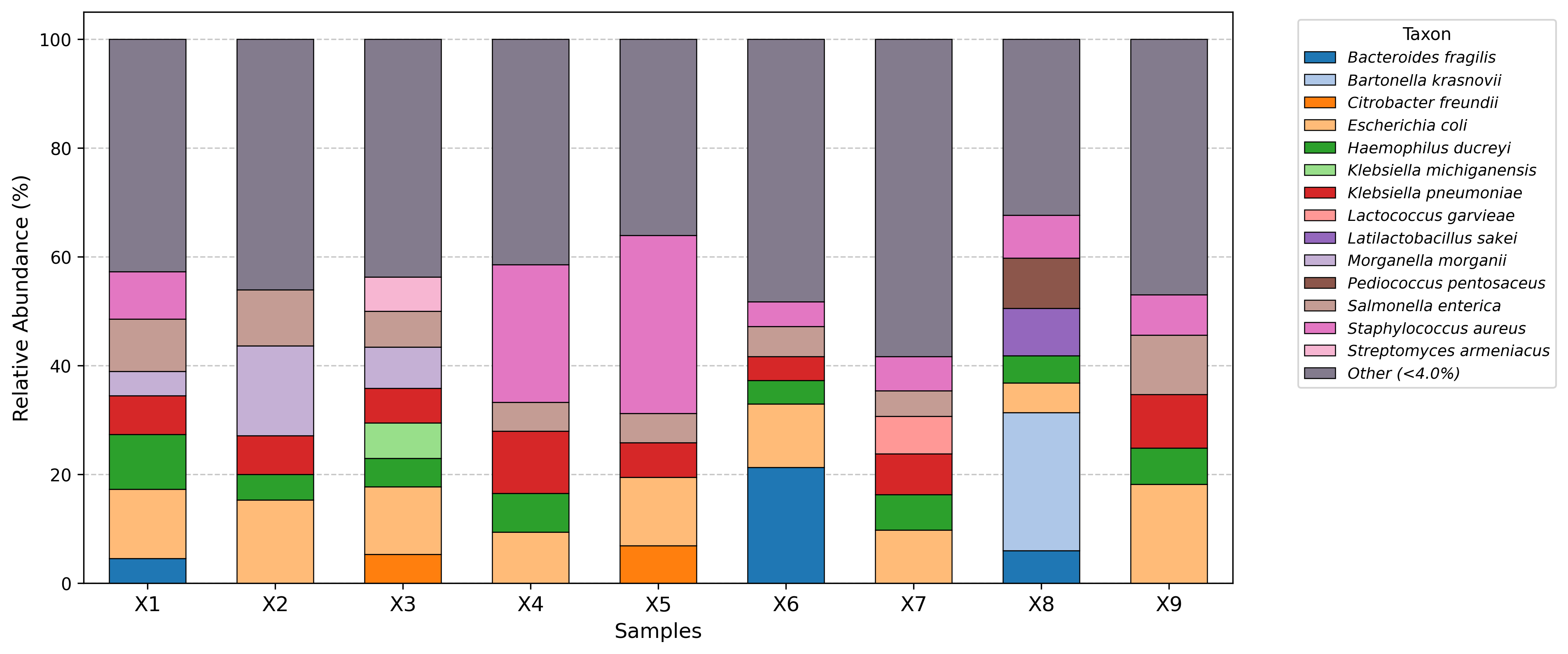 |
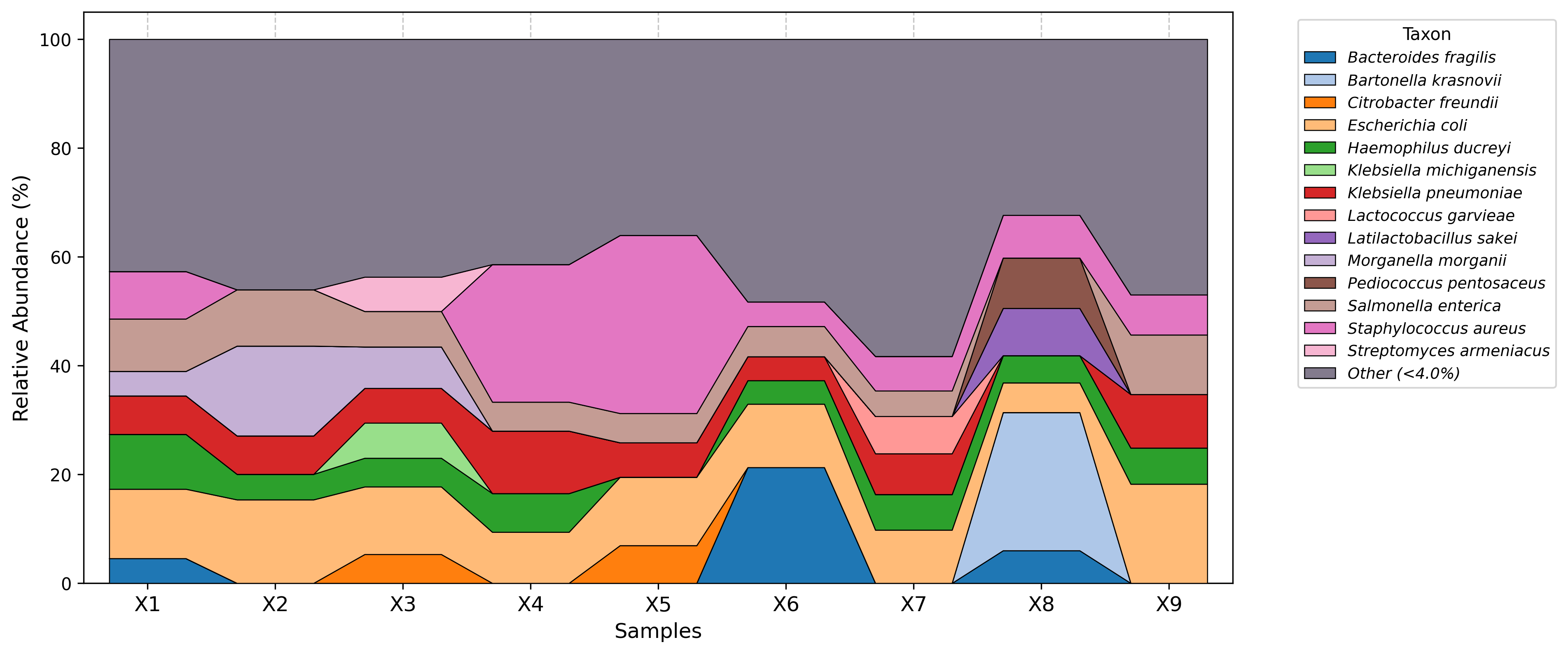 |
| Stacked Barplot + Streamgraph | Clustermap |
|---|---|
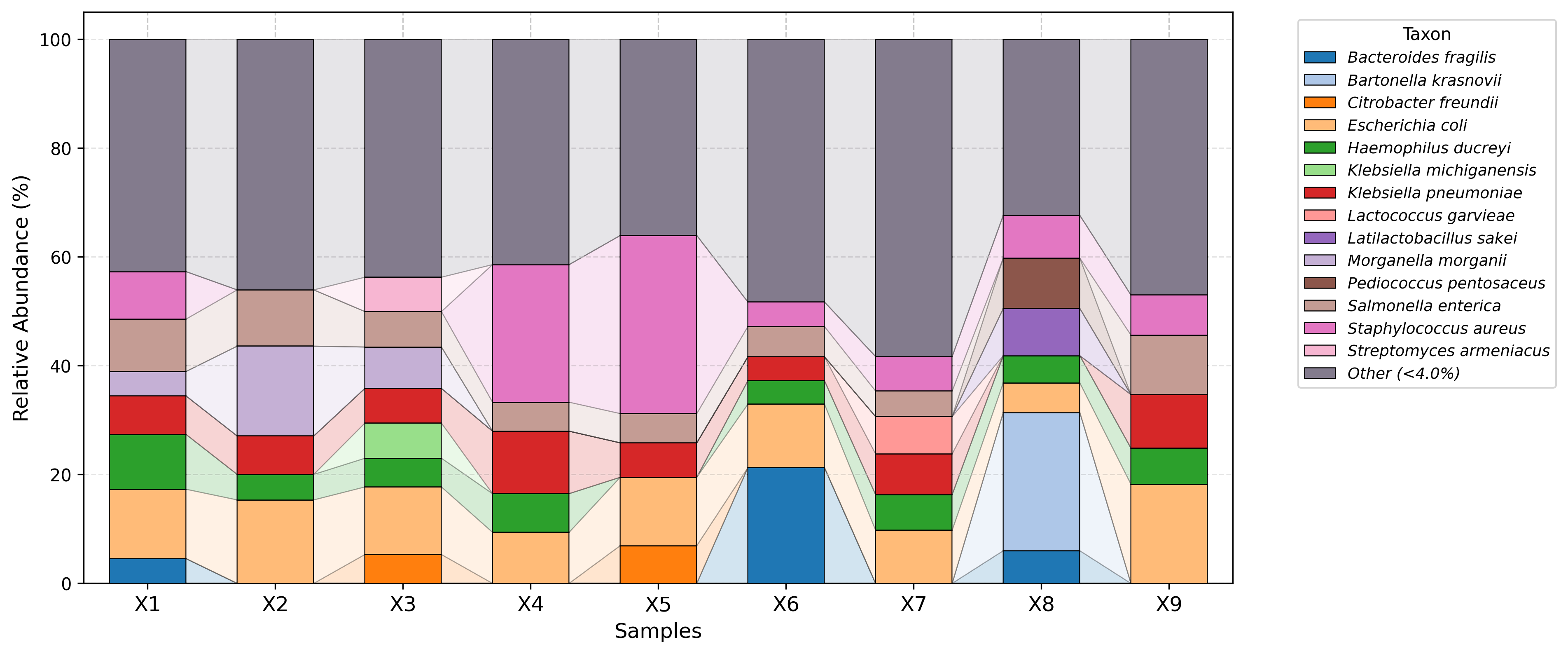 |
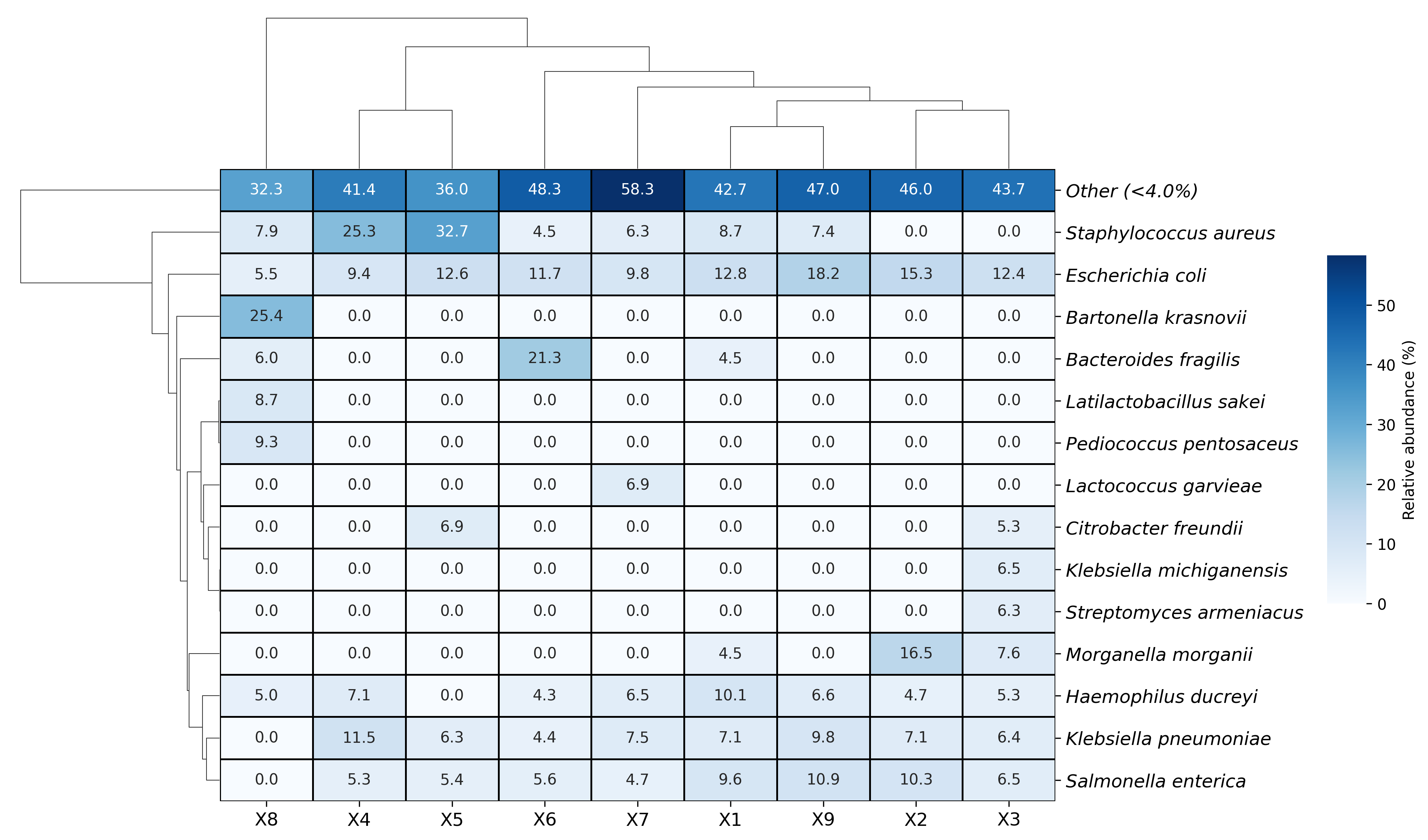 |
Example Output Structure
After running the full pipeline, the output directory will look like this:
results/
├─ counts/ # Total abundance CSV output
│ ├─ counts_species.csv
│ ├─ counts_genus.csv
│ ├─ ...
│ └─ counts_phylum.csv
├─ rel_abund/ # Relative abundance CSV output
│ ├─ ra_species.csv
│ ├─ ra_genus.csv
│ ├─ ...
│ └─ ra_phylum.csv
├─ diversity/ # Diversity metrics
│ ├─ alpha_div.csv
│ ├─ beta_div_bray.csv
│ └─ beta_div_jaccard.csv
├─ intermediate/ # Intermediate files
│ ├─ mpa/ # Converted MPA files
│ │ ├─ {sample}.txt
│ │ ├─ ...
│ ├─ COMBINED.txt # Merged MPA table
│ └─ txt/ # Extracted taxonomic levels in TXT
│ ├─ counts_species.txt
│ ├─ counts_genus.txt
│ ├─ ...
│ └─ counts_phylum.txt
└─ krakenparser.log # Pipeline execution logs
Conclusion
KrakenParser provides a simple and automated way to convert Kraken2 reports into usable CSV files for downstream analysis. You can run the full pipeline with a single command or use individual modules as needed.
For any issues or feature requests, feel free to open an issue on GitHub!
🚀 Happy analyzing!
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file krakenparser-1.1.2.tar.gz.
File metadata
- Download URL: krakenparser-1.1.2.tar.gz
- Upload date:
- Size: 49.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
7328beb4811d7161d3b1a2e107dbc3454f9f9547561f6980cc28ea78d217103b
|
|
| MD5 |
71001ca320704f9c365d3bb6596a3942
|
|
| BLAKE2b-256 |
b9ba4d9533a251b0c71146d579df3272eb62f50387f18c0c18f0188e96ed38f5
|
Provenance
The following attestation bundles were made for krakenparser-1.1.2.tar.gz:
Publisher:
publish.yml on PopovIILab/KrakenParser
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
krakenparser-1.1.2.tar.gz -
Subject digest:
7328beb4811d7161d3b1a2e107dbc3454f9f9547561f6980cc28ea78d217103b - Sigstore transparency entry: 1717614110
- Sigstore integration time:
-
Permalink:
PopovIILab/KrakenParser@6b479be45bf1cf233c3aad0ba1e4b43af760660c -
Branch / Tag:
refs/tags/v.1.1.2 - Owner: https://github.com/PopovIILab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@6b479be45bf1cf233c3aad0ba1e4b43af760660c -
Trigger Event:
push
-
Statement type:
File details
Details for the file krakenparser-1.1.2-py3-none-any.whl.
File metadata
- Download URL: krakenparser-1.1.2-py3-none-any.whl
- Upload date:
- Size: 60.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1d8dc8e5500e354a842ebcfcd9b7cb2a79a193eb98de6fde00ed7897818e6470
|
|
| MD5 |
a699f88eeb161f2326d4585f4bb48fe7
|
|
| BLAKE2b-256 |
cd6903cf792cafac218dc4e31285ac21878ae4b604253b56eb5643aef8fac972
|
Provenance
The following attestation bundles were made for krakenparser-1.1.2-py3-none-any.whl:
Publisher:
publish.yml on PopovIILab/KrakenParser
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
krakenparser-1.1.2-py3-none-any.whl -
Subject digest:
1d8dc8e5500e354a842ebcfcd9b7cb2a79a193eb98de6fde00ed7897818e6470 - Sigstore transparency entry: 1717614201
- Sigstore integration time:
-
Permalink:
PopovIILab/KrakenParser@6b479be45bf1cf233c3aad0ba1e4b43af760660c -
Branch / Tag:
refs/tags/v.1.1.2 - Owner: https://github.com/PopovIILab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@6b479be45bf1cf233c3aad0ba1e4b43af760660c -
Trigger Event:
push
-
Statement type:







