No project description provided

Project description
meiosim
meiosim provides a single, lightweight data structure that bundles genotypes, individual metadata and a genetic map together. It includes the operations breeders and quantitative geneticists reach for most often:
- basic data manipulation,
- simulating meiosis and crosses,
- a ready-to-use Arabidopsis thaliana panel.
Installation
Install meiosim with pip:
pip install meiosim
Quick start
from meiosim import Arabidopsis
import matplotlib.pyplot as plt
# Load 5,000 random SNPs from the 1001 Genomes panel
pop = Arabidopsis(n_SNPs = 5000, seed = 42)
# Visualise population structure
plt.figure(figsize=(12,6))
pop.plot(group="country")
plt.show()
Core concepts
Everything in meiosim revolves around the Population object, which holds three
aligned pieces of data:
| Attribute | Type | Shape | Description |
|---|---|---|---|
genotypes |
np.ndarray |
(n_individuals, n_markers) |
Allele dosage coded -1 / 0 / 1; missing values are np.nan. |
metadata |
pd.DataFrame |
n_individuals rows |
One row per individual. Columns are free-form. |
map |
pd.DataFrame |
n_markers rows |
Marker map; needs at least chromosome and cM columns. |
On construction, meiosim guarantees three metadata columns:
individual: a primary key for each individual, automatically created as a 16-character SHA-256 fingerprint if absent.sire/dam: parental identifiers, filled in by crossing operations.
Optional attributes appear once you run the relevant method:
phases: a list[hap0, hap1]of two haplotype matrices, created byphasing()and propagated through crossing.phenotypes: apd.DataFrameof trait values.
A shared numpy random generator (rng) makes every stochastic operation reproducible.
The Population API
Constructor
pop = Population(genotypes, metadata, map, seed = 42)
Build a population directly from SNP data. genotypes is a -1/0/1 matrix
(np.nan allowed), metadata and map are DataFrames aligned to the rows and
columns of genotypes respectively.
merge(*populations) (classmethod)
combined = Population.merge(pop_a, pop_b, pop_c)
Stack two or more populations that share an identical map. Metadata is concatenated, genotypes are row-stacked, and phases are merged when all input populations carry them.
subset(on)
Filter individuals in place, on either:
dict: match metadata columns, e.g.{"country": "SWE"}or{"country": ["SWE", "FIN"], "stage": "line"}. Multiple keys are combined withand; multiple values per key withor.list[str]: keep individuals by theirindividualID.list[int]: keep individuals by row index.
pop.subset({"country": "SWE"}).subset(list(range(20)))
split(size)
Randomly partition the markers into two new populations of size and
n_markers - size columns:
snp, qtl = pop.split(2_000) # snp has 2,000 markers, qtl gets the rest
Each individual is preserved in both halves; phases are split alongside.
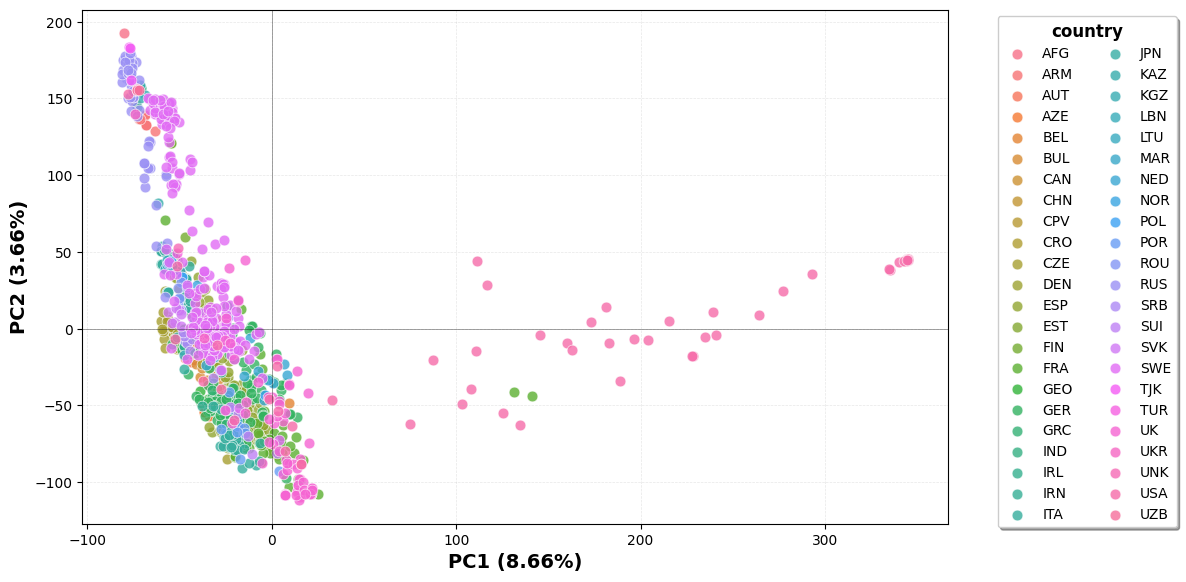
plot(group=None)
PCA scatter of the population on the first two principal components (genotypes
are mean-imputed first). Pass a metadata column name to group to colour points
by category (a group named "other" is drawn in grey).
pop.plot(group="country")
phasing()
Infer haplotypes and populate self.phases.
⚠️ Designed for fully inbred material: for an actual phasing of heterozygous material, an external tool must be used.
cross(parents=None, selfing=True, n_progeny=1, n_cores=5)
Generate progeny from all pairwise combinations of the chosen parents.
parents— a list of IDs (str) or indices (int), or a single one. Defaults to every individual in the population.selfing— ifTrue, self-crosses are allowed (pairs withp1 <= p2); ifFalse, only distinct pairs are crossed (p1 < p2).n_progeny— offspring produced per cross.n_cores— workers used by the underlying meiosis.
Returns a new Population whose metadata is a pedigree (individual, sire,
dam) and whose phases carry the two inherited gametes.
f1 = founders.cross(selfing=False) # all distinct pairwise crosses
selfing(individual, n_generations, n_cores=5)
Repeatedly self-fertilise one individual to drive it towards homozygosity,
returning the population after n_generations rounds. The original parents are
carried over into the resulting metadata.
line = f1.selfing(id1, n_generations=6)
Multiprocessing
cross, selfing and meiosis use a
ProcessPoolExecutor. On macOS/Windows, set the start method to 'fork'
(where supported) before launching workers, as shown in the example, and run
inside an if __name__ == "__main__": guard.
Arabidopsis dataset
The 1001 genome collection of A. thaliana is provided as a resource.
The Arabidopsis constructor base loader reads SNPs, accessions,
positions, derives a genetic map and provides phenotypes.
Optionally, down-samples to n_SNPs random markers (n_SNPs=0 keeps them all).
The Arabidopsis data come from the following resources:
Base loader for an A. thaliana SNP matrix. Reads SNPs, accessions and positions, joins the master accession list as metadata,
| Feature | Reference |
|---|---|
| 1001 Arabidopsis genomes | The 1001 Genomes Consortium (2016). 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis thaliana. Cell. |
| 1001 Arabidopsis phenotypes | Grimm et al. (2017). easyGWAS: A Cloud-Based Platform for Comparing the Results of Genome-Wide Association Studies. The Plant Cell. |
| Genetic map | Salomé et al. (2012). The recombination landscape in Arabidopsis thaliana F2 populations. Heredity. |
| RegMap SNP panel | Horton et al. (2012). Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nature Genetics. |
| RegMap SNP panel | Pisupati et al. (2017) - Verification of Arabidopsis stock collections using SNPmatch, a tool for genotyping high-plexed samples |
Citation
Please cite this package as:
Marchal, A., & Raimondi, D. (2026). meiosim - a toolbox to simulate crosses and play with quantitative genetics in Python [Computer software]. Zenodo. https://doi.org/10.5281/zenodo.21134827
Code
The code is available at this address.
License
Copyright © 2026 CNRS, University of Montpellier
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program. If not, see https://www.gnu.org/licenses/.
Release history Release notifications | RSS feed

Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file meiosim-0.1.0.tar.gz.
File metadata
- Download URL: meiosim-0.1.0.tar.gz
- Upload date:
- Size: 29.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
6c453a0760b80127b6b73a3ce5947d5db34520dddd0f7ea4686abd8f0cdc71a0
|
|
| MD5 |
3208edd22976f25a8bd7dd48a9aa392c
|
|
| BLAKE2b-256 |
e728a7eac176ce885b1ef71c24f1efca92f710c1c6469ea645dbffaffe67bd70
|
File details
Details for the file meiosim-0.1.0-py3-none-any.whl.
File metadata
- Download URL: meiosim-0.1.0-py3-none-any.whl
- Upload date:
- Size: 29.3 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
59a113c2577a345ca15fa93651b4a98e244cadbb478f76dae7a679f2d3cb666a
|
|
| MD5 |
4135986cae8502ebee36fb23fc5a5f39
|
|
| BLAKE2b-256 |
fe827e96f6e6146d1a5ed214a85c2191854bbb49d59a7a07c64f77ef5f05ba1a
|







