A Python package for visualizing protein-ligand interactions with 2D ligand structure representation

Project description
PandaMap: A Python Package for Visualizing Protein–Ligand Interactions
Protein AND ligAnd interaction MAPper — comprehensive detection, visualization, and empirical binding affinity estimation for protein–ligand complexes.







What's New in v4.2
| Feature | Description |
|---|---|
| Scientifically validated cutoffs | H-bond lower bound 2.5 Å, ionic/salt bridge 5.5 Å, halogen lower bound 2.5 Å — aligned with PLIP and crystallographic surveys |
| RDKit 2D coordinates | Chemically accurate 2D ligand layout when RDKit is available; PCA projection fallback requires no extra dependencies |
| Topology-based ring detection | Iterative leaf-node pruning on the bond graph — identifies rings of any size without SMILES |
| Exact aromatic atom filtering | Per-residue ring-atom name sets (PHE/TYR/TRP/HIS) eliminate false π-system contacts from β-carbons |
| Improved ΔG estimation | Per-residue deduplication + distance decay + rotatable bond entropy penalty; thermodynamically calibrated Kd labels at 298 K |
| Trajectory analysis | Multi-frame PDB/NMR ensemble analysis with per-interaction occupancy statistics and CSV export |
| Arc stagger in 2D diagram | Multiple interactions to the same residue fan out with alternating curvature — no marker overlap |
--deltaG CLI flag |
Estimate binding free energy directly from the command line |
Features
- 16 interaction classes detected with crystallographically validated distance thresholds:
- Hydrogen bonds (2.5–3.5 Å, N/O pairs)
- π–π stacking, cation–π, pi–cation, carbon–π, donor–π, amide–π, alkyl–π
- Hydrophobic contacts (4.0 Å)
- Ionic interactions and salt bridges (5.5 Å)
- Halogen bonds (2.5–3.5 Å)
- Metal coordination (2.8 Å)
- Covalent bonds (≤2.1 Å, CYS/SER/LYS/HIS)
- Attractive and repulsive charge interactions
- Three output formats: 2D PNG diagram, interactive 3D HTML (3Dmol.js), plain-text report
- Empirical ΔG scoring with per-residue deduplication, distance decay, and rotor penalty
- Multi-frame trajectory analysis with occupancy statistics and CSV export
- Solvent accessibility: DSSP (preferred) → Shrake–Rupley fallback → geometric fallback
- Multi-format input: PDB, mmCIF/CIF, PDBQT (AutoDock Vina)
Installation
pip install pandamap
Optional dependencies
pip install pandamap[fancy] # coloured CLI output (rich)
pip install pandamap[viz] # programmatic 3D viewer (py3Dmol)
pip install pandamap[full] # all extras
External optional: DSSP (accurate solvent accessibility)
brew install dssp # macOS
sudo apt-get install dssp # Linux
# Windows: https://swift.cmbi.umcn.nl/gv/dssp/
RDKit (for chemically accurate 2D ligand coordinates):
conda install -c conda-forge rdkit # recommended
pip install rdkit # pip alternative
PandaMap works without RDKit — it falls back to PCA-based 2D projection automatically.
Quick Start
# 2D interaction diagram
pandamap structure.pdb
# Specify ligand, generate report and 3D viewer
pandamap complex.pdb --ligand PFL --report --3d
# Estimate binding free energy
pandamap complex.pdb --ligand PFL --deltaG
# Full analysis
pandamap complex.pdb --ligand LIG --report --3d --deltaG --dpi 300
Command-Line Reference
pandamap <structure_file> [options]
Positional arguments:
structure_file Path to PDB, mmCIF/CIF, or PDBQT file
Options:
-l, --ligand NAME Three-letter residue code of the ligand (default: auto-detect)
-o, --output FILE Output PNG file path
-r, --report Generate plain-text interaction report
--report-file FILE Path for text report
--3d Generate interactive 3D HTML visualization
--3d-output FILE Path for 3D HTML file
--deltaG Estimate binding free energy (ΔG, kcal/mol)
--dpi DPI Output PNG resolution (default: 300)
-t, --title TEXT Custom diagram title
--width PX 3D viewer width in pixels (default: 800)
--height PX 3D viewer height in pixels (default: 600)
--no-surface Hide protein surface in 3D viewer
--no-3d-cues Disable depth cues in 2D diagram
-v, --version Show version
-h, --help Show help
Python API
Single-structure analysis
from pandamap import HybridProtLigMapper
mapper = HybridProtLigMapper("complex.pdb", ligand_resname="LIG")
mapper.detect_interactions()
# 2D diagram
mapper.visualize(output_file="interactions.png")
# Text report
from pandamap.improved_interaction_detection import ImprovedInteractionDetection
detector = ImprovedInteractionDetection()
detector.generate_report(
ligand_metadata={
'hetid': mapper.ligand_residue.resname,
'chain': mapper.ligand_residue.parent.id,
'position': mapper.ligand_residue.id[1],
'longname': mapper.ligand_residue.resname,
'type': 'LIGAND',
},
interaction_data=mapper.interactions,
output_file="report.txt"
)
# Inspect raw interactions
for itype, contacts in mapper.interactions.items():
if contacts:
print(f"{itype}: {len(contacts)} contacts")
Empirical ΔG estimation
result = mapper.estimate_binding_affinity()
print(f"ΔG ≈ {result['dG_estimated']:.2f} kcal/mol")
print(result['interpretation'])
print(result['note'])
for itype, info in result['breakdown'].items():
print(f" {itype} (n={info['unique_residues']}): {info['contribution_kcal_mol']:+.2f} kcal/mol")
ΔG interpretation (298 K, ΔG = −RT·ln Kd):
| ΔG (kcal/mol) | Kd range | Label |
|---|---|---|
| < −12 | ~nM or better | Very strong binder |
| −9 to −12 | nM–µM | Strong binder |
| −6 to −9 | µM | Moderate binder |
| −3 to −6 | mM | Weak binder |
| ≥ −3 | — | Very weak / no binding |
Note: Empirical estimate ±2–3 kcal/mol. Not a substitute for FEP or MM-GBSA.
3D visualization
from pandamap.create_3d_view import create_pandamap_3d_viz
create_pandamap_3d_viz(
mapper=mapper,
output_file="interactions_3d.html",
width=1024,
height=768,
show_surface=True
)
Multi-frame trajectory analysis
from pandamap import analyze_trajectory
summary = analyze_trajectory(
trajectory_file="simulation.pdb", # multi-MODEL PDB
ligand_resname="LIG",
output_dir="./trajectory_output",
visualize_frames=False # set True to generate per-frame PNGs
)
print(f"Frames analysed: {summary['n_frames']}")
print(f"Mean ΔG: {summary['mean_dG']:.2f} ± {summary['std_dG']:.2f} kcal/mol")
# Per-residue occupancy CSV written to ./trajectory_output/trajectory_analysis.csv
Distance Cutoffs
All cutoffs are validated against PLIP and published crystallographic surveys:
| Interaction | Cutoff | Reference |
|---|---|---|
| Hydrogen bond | 2.5–3.5 Å | PLIP; Auffinger 2004 |
| π–π stacking | 5.5 Å (atom–atom) | McGaughey 1998 |
| Hydrophobic | 4.0 Å | Bissantz 2010 |
| Ionic / salt bridge | 5.5 Å | Kumar & Nussinov 1999 |
| Halogen bond | 2.5–3.5 Å | Auffinger 2004 |
| Metal coordination | 2.8 Å | CSD surveys |
| Covalent | ≤2.1 Å | — |
| Repulsion | 4.0 Å | — |
Example Outputs
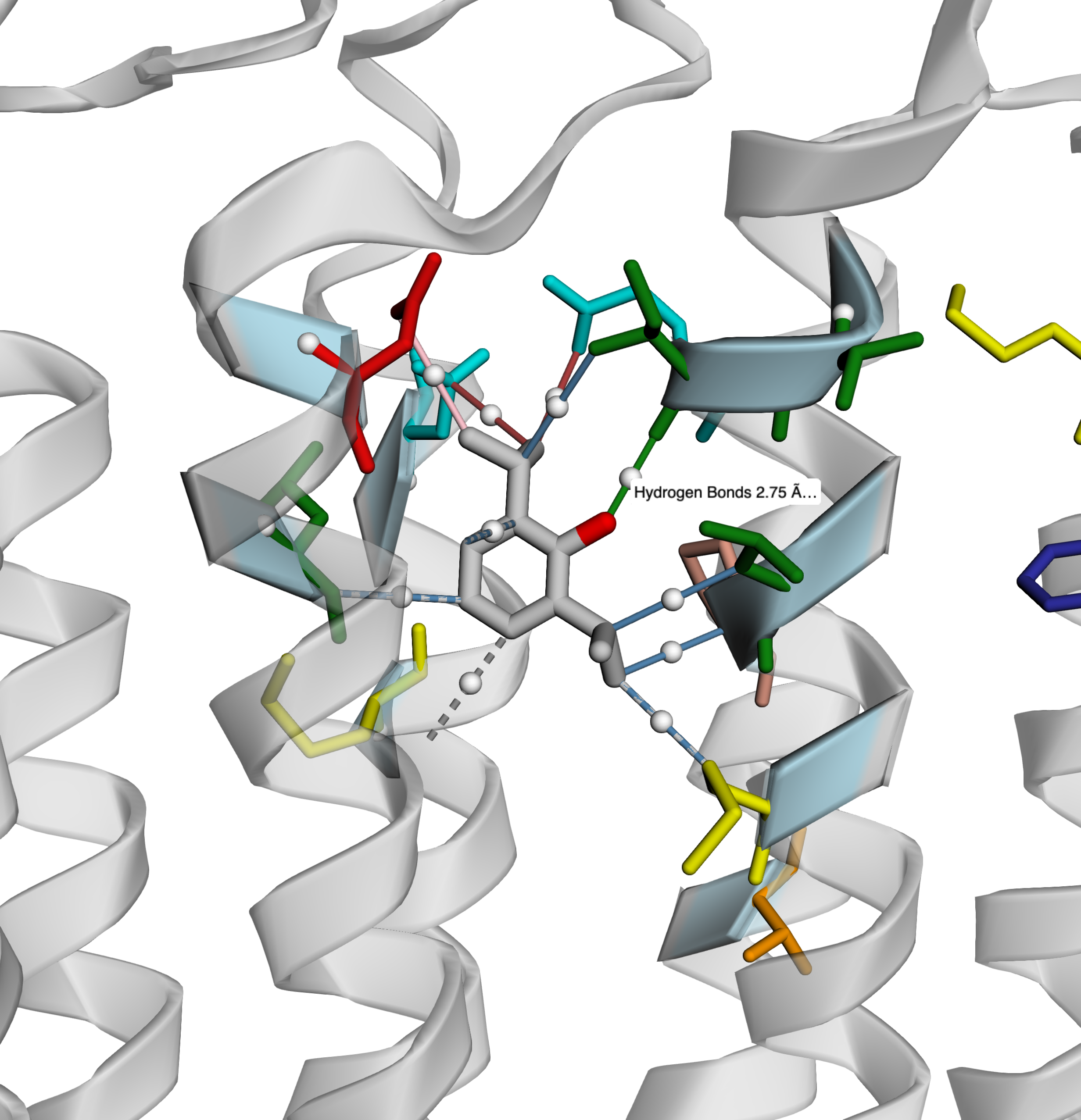
2D Interaction Diagram
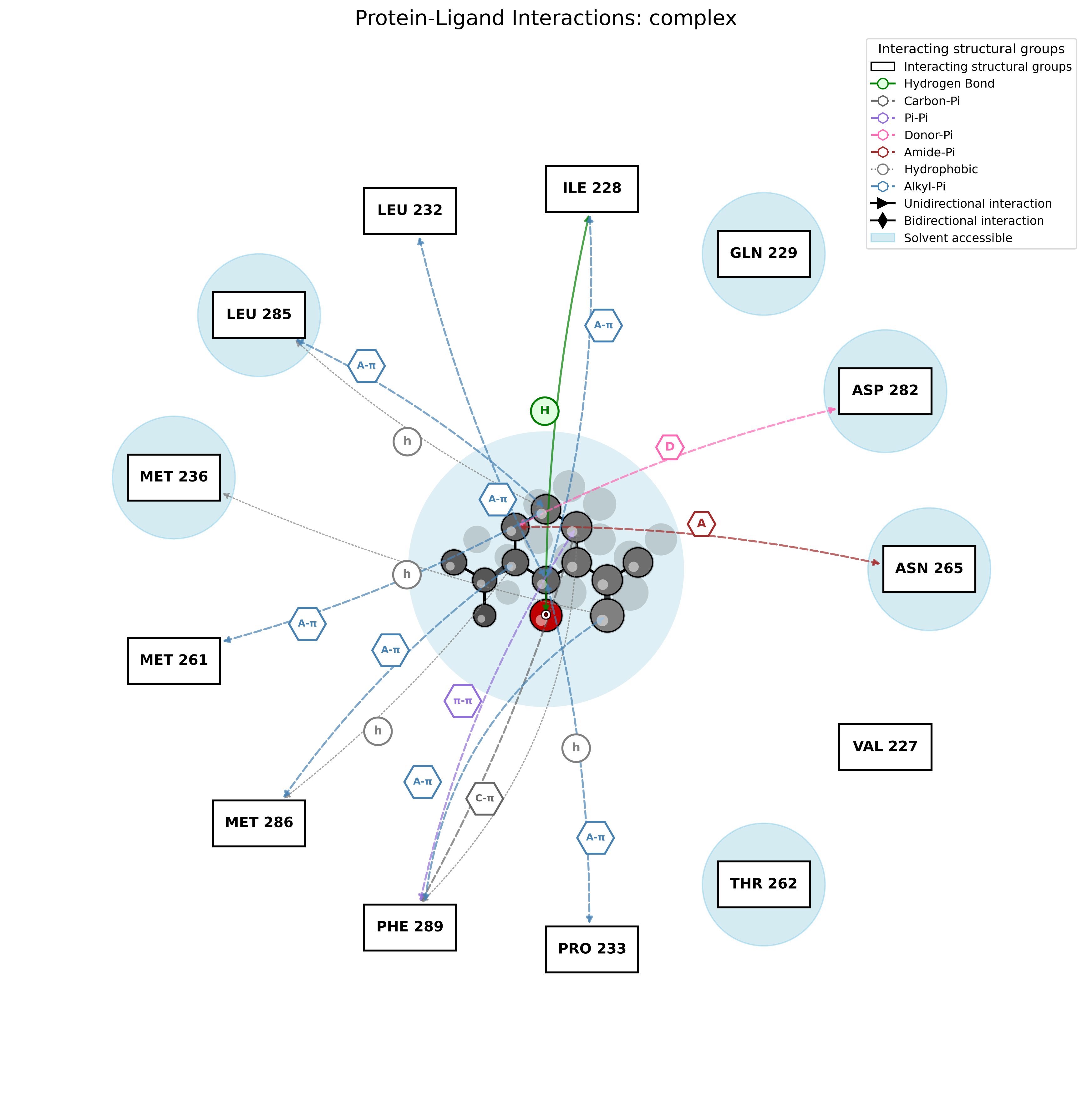
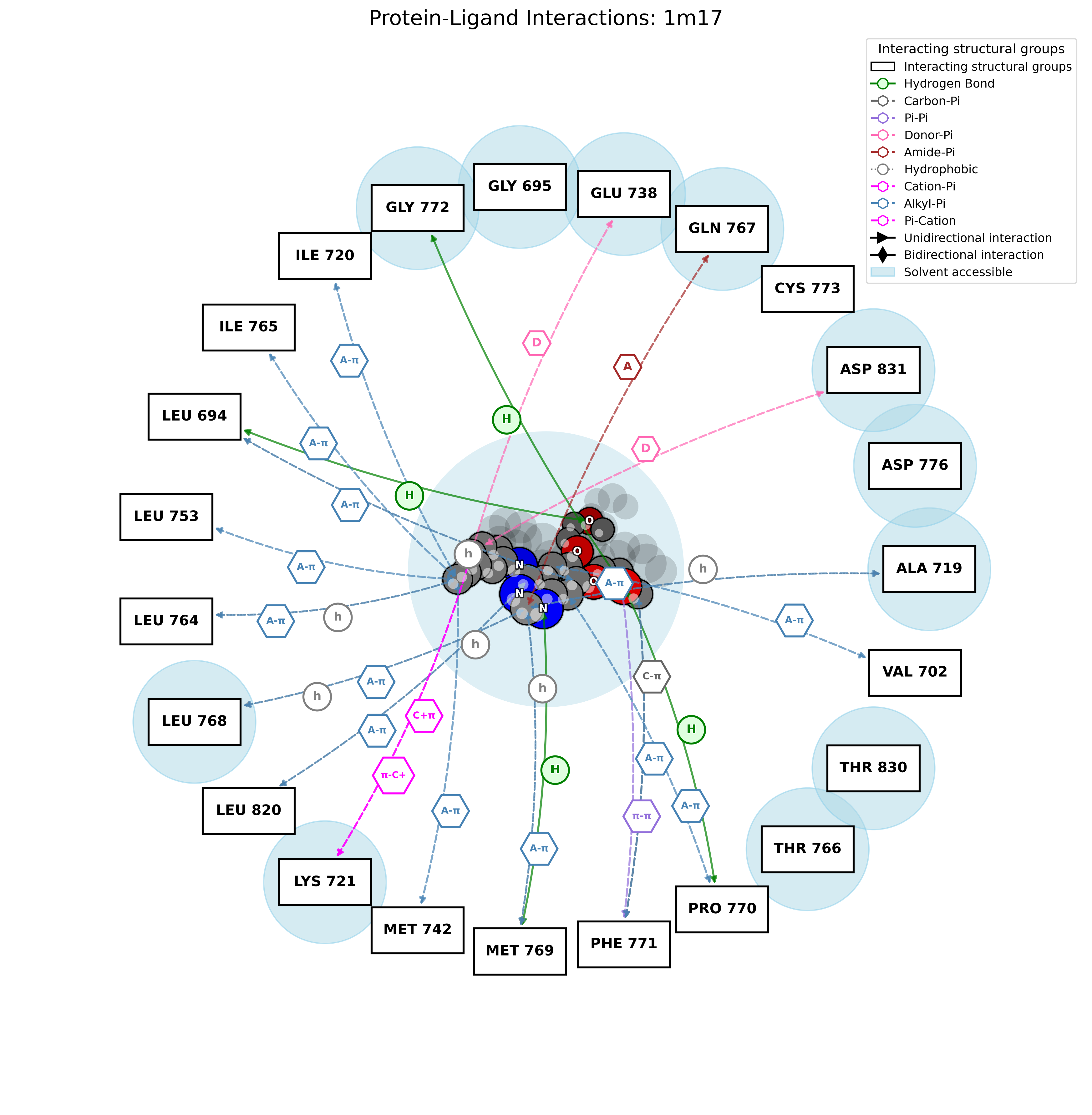
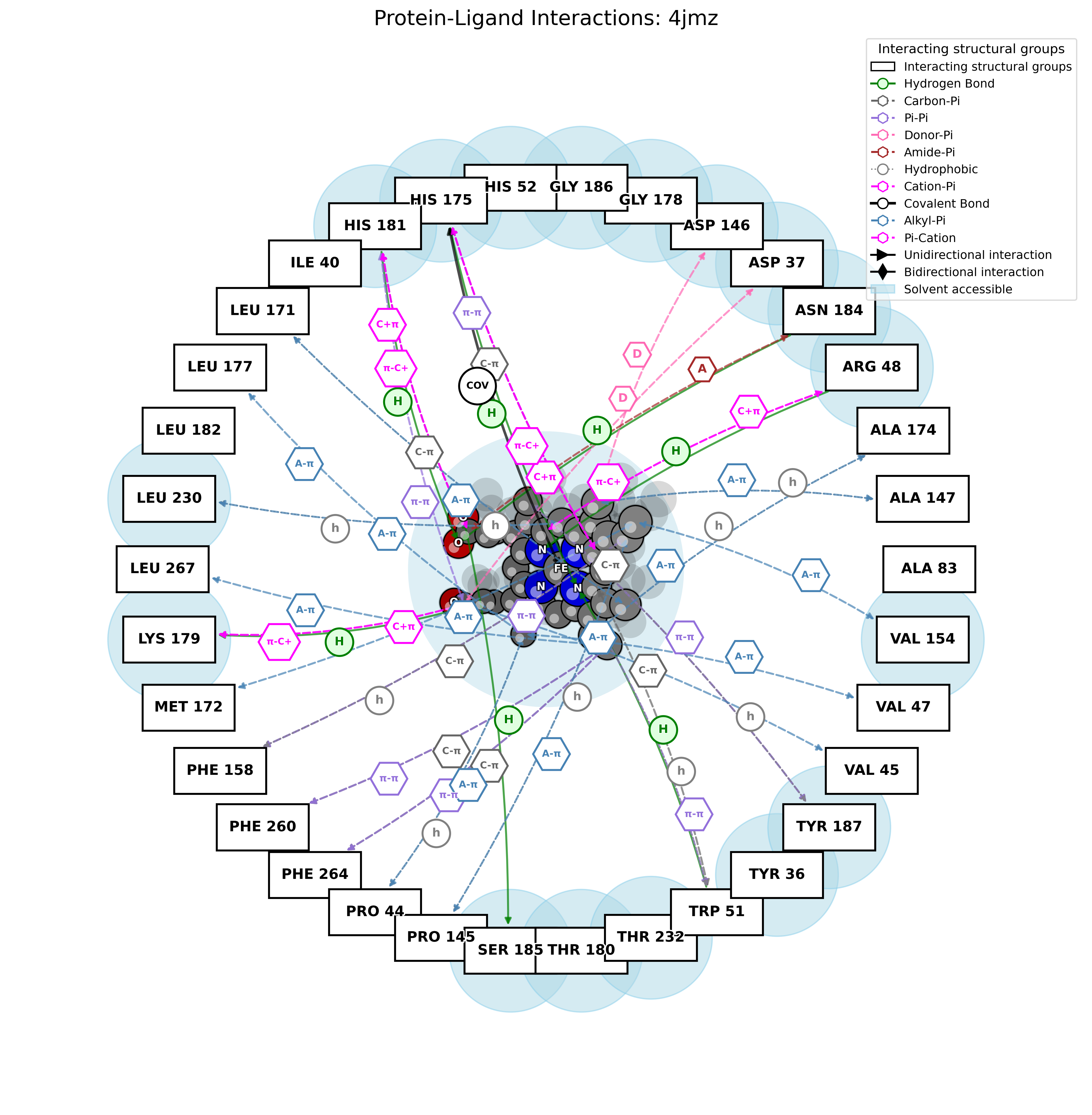
Text Report
=============================================================================
PandaMap Interaction Report
=============================================================================
Ligand: PAH:A:439
Name: PAH
Type: LIGAND
Interacting Chains: A
Interacting Residues: 13
Interaction Summary:
Hydrogen Bonds: 10
Carbon-π Interactions: 1
Metal Coordination: 4
Ionic Interactions: 2
Salt Bridges: 2
Alkyl-π Interactions: 1
Attractive Charge: 2
Repulsion: 5
Hydrogen Bonds:
1. GLU168A -- 2.66Å -- PAH
2. ASP246A -- 2.60Å -- PAH
3. GLN167A -- 3.10Å -- PAH
4. ASP320A -- 3.46Å -- PAH
5. LYS396A -- 3.05Å -- PAH
...
=============================================================================
ΔG Estimation Output
--- Estimated Binding Affinity ---
ΔG ≈ -7.42 kcal/mol
Strong binder (Kd ~nM–µM range)
Breakdown:
hydrogen_bonds (n=10): -8.63 kcal/mol
metal_coordination (n=4): -6.80 kcal/mol
ionic (n=2): -2.91 kcal/mol
hydrophobic (n=3): -0.72 kcal/mol
rotatable_bond_penalty (n=2): +1.00 kcal/mol
----------------------------------
Citation
If you use PandaMap in your research, please cite:
Pritam Kumar Panda. (2025). PandaMap: A Python Package for Comprehensive
Visualization of Protein–Ligand Interaction Networks and Empirical Binding
Affinity Estimation. Stanford University, CA, USA
https://github.com/pritampanda15/PandaMap
License
MIT License — see LICENSE for details.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file pandamap-4.2.1.tar.gz.
File metadata
- Download URL: pandamap-4.2.1.tar.gz
- Upload date:
- Size: 52.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
42f1fde77c81984e13946fbd4f178ba7e4a7b5922762e3cca5338947cc25f0ae
|
|
| MD5 |
a3229ed72c3eaba9e38dde2e31e3b7f8
|
|
| BLAKE2b-256 |
163a4c9aefc8abb23ecd17ba032bb393cc1be7eb78f799da38def92387426bff
|
File details
Details for the file pandamap-4.2.1-py3-none-any.whl.
File metadata
- Download URL: pandamap-4.2.1-py3-none-any.whl
- Upload date:
- Size: 50.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9ad623afc300b161621bbc7a1890f1f80c9431a20c4e24ba7661f543da598af4
|
|
| MD5 |
d4e2eb943e99eadcb2d5a7b3df07be59
|
|
| BLAKE2b-256 |
ac65467d1b15da4eb477bb73f8cd787a17203e9f79d6757ca84bb4c8ae620874
|







