For matching species databases

Project description
Phylo-Match
Phylo-Match is a package for correcting misspellings or disparate labelings in phylogenetic trees
Installation
This project was built with python v3.8 (python3). Later versions should be fine, but no guarantees for earlier. Not compatible with python 2.
Check that you have python3 installed:
python3 --version
Output should be something like:
Python 3.7.1
If --version does not return a version, follow Python3 installation instructions (or install Python3 your own way)
Use the package manager pip3 to install phylo-match.
Install pip:
python3 -m ensurepip --upgrade
Install Phylo-Match:
pip3 install phylo-match
Upgrade Phylo-Match:
pip install phylo-match -U
Usage
phylo-match
Use the gui to select a database file (.csv), and a taxa tree (.nexus) to match the database to. Enter the number of your species column in the box, if the taxa you are matching are not in the first column (index counts from 0, so enter 0 for first column, 1 for second, etc.)
Click run when you are happy with your selection.
Phylo-Match does all of its calculations and api requests upfront, so users may have to wait 10-15 minutes after run is clicked, depending on internet speed and whether these taxa are already in their local cache.
This time can be minimized by unchecking 'Lookup Taxa Info' - a good idea if you're very familiar with the taxa, but the project will not provide information about matches beyond the name.
Information about the DB's taxa will be on the left-hand side. All similar entries in the .nexus file will appear in the middle of the screen. Click on the name you'd like to change the entry to, manually enter a name on the bottom, or click on 'same species', or 'same genus' for additional options, if available.
Once all selections have been made, a new .csv file will be created in the same directory as the original database .csv file.
Your matching progress is not saved until a new file is created, so be prepared to start over if you exit halfway through.
Downloaded content will cache immediately upon download, so starting over will take significantly less time.
Examples:
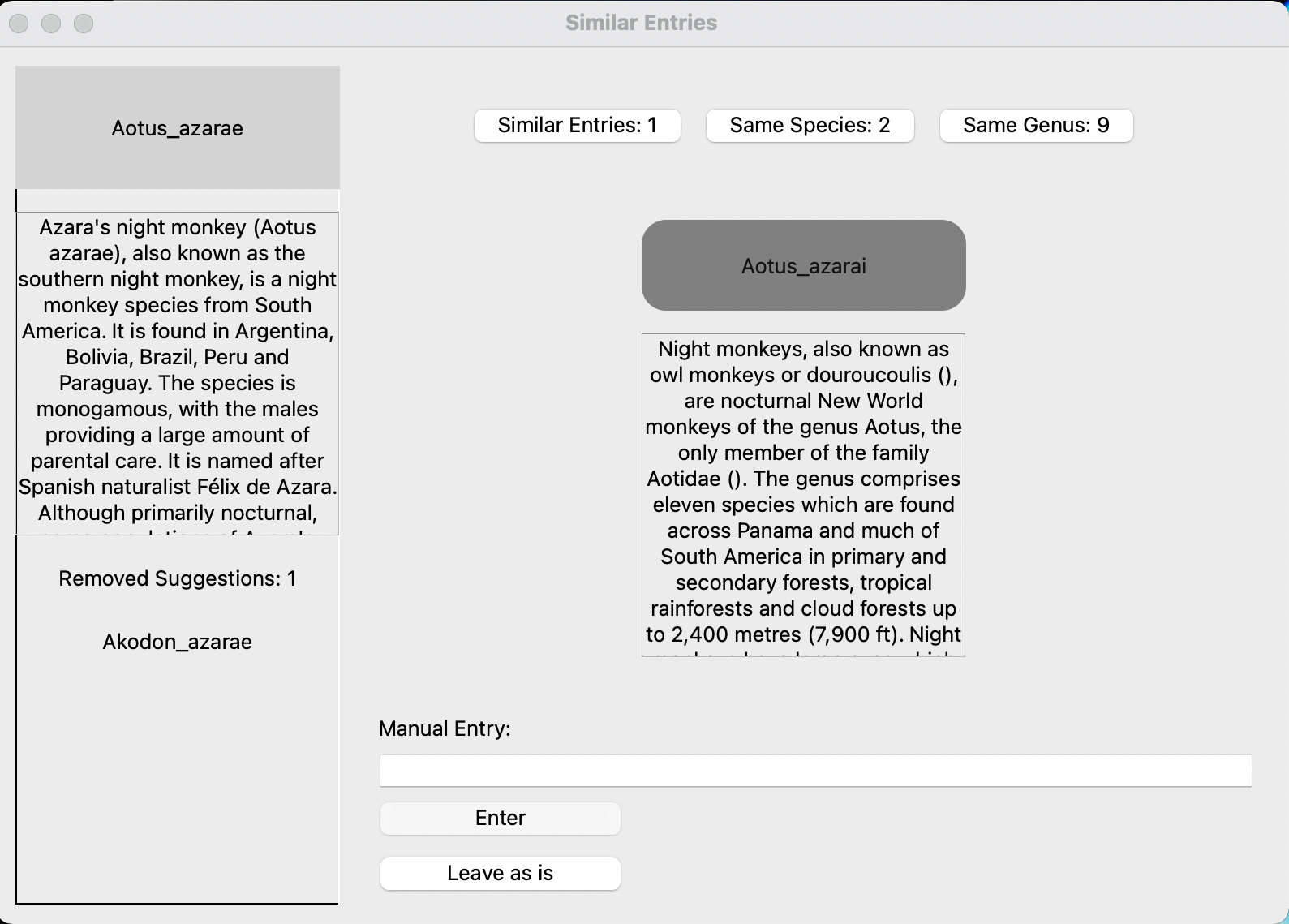
The program (correctly) thinks my best bet to match the database taxon Aotus azarae is Aotus azarai from the tree. But if I don't like that option I can click on 'same species' to see other taxa in the tree with the species name "azarae" or 'same genus' to see other members of the genus Aotus.
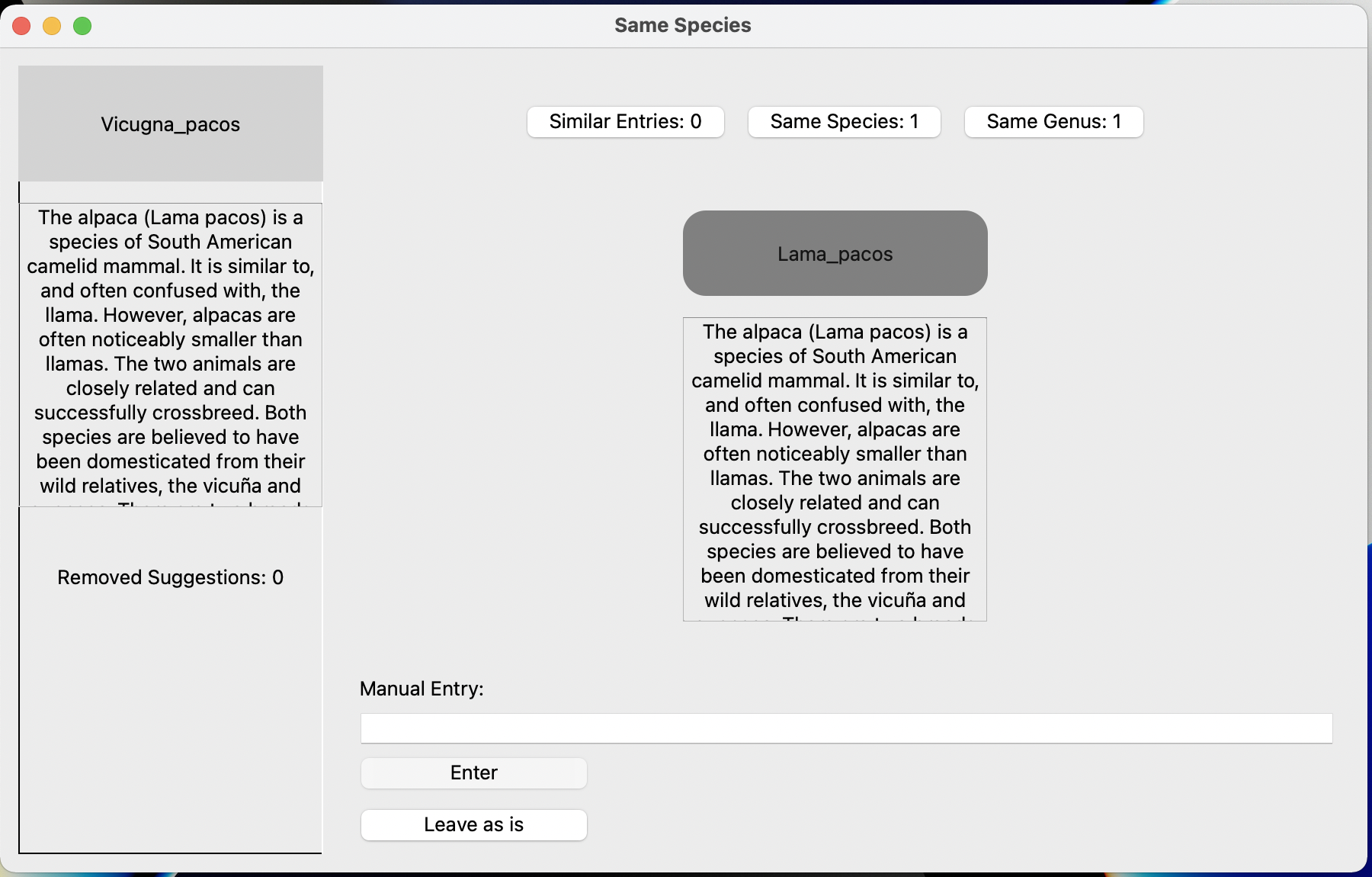
Here's an example of the 'same species' option at work: The database has Vicunga pacos but the tree has Lama pacos. This is useful if the genus has been split up.
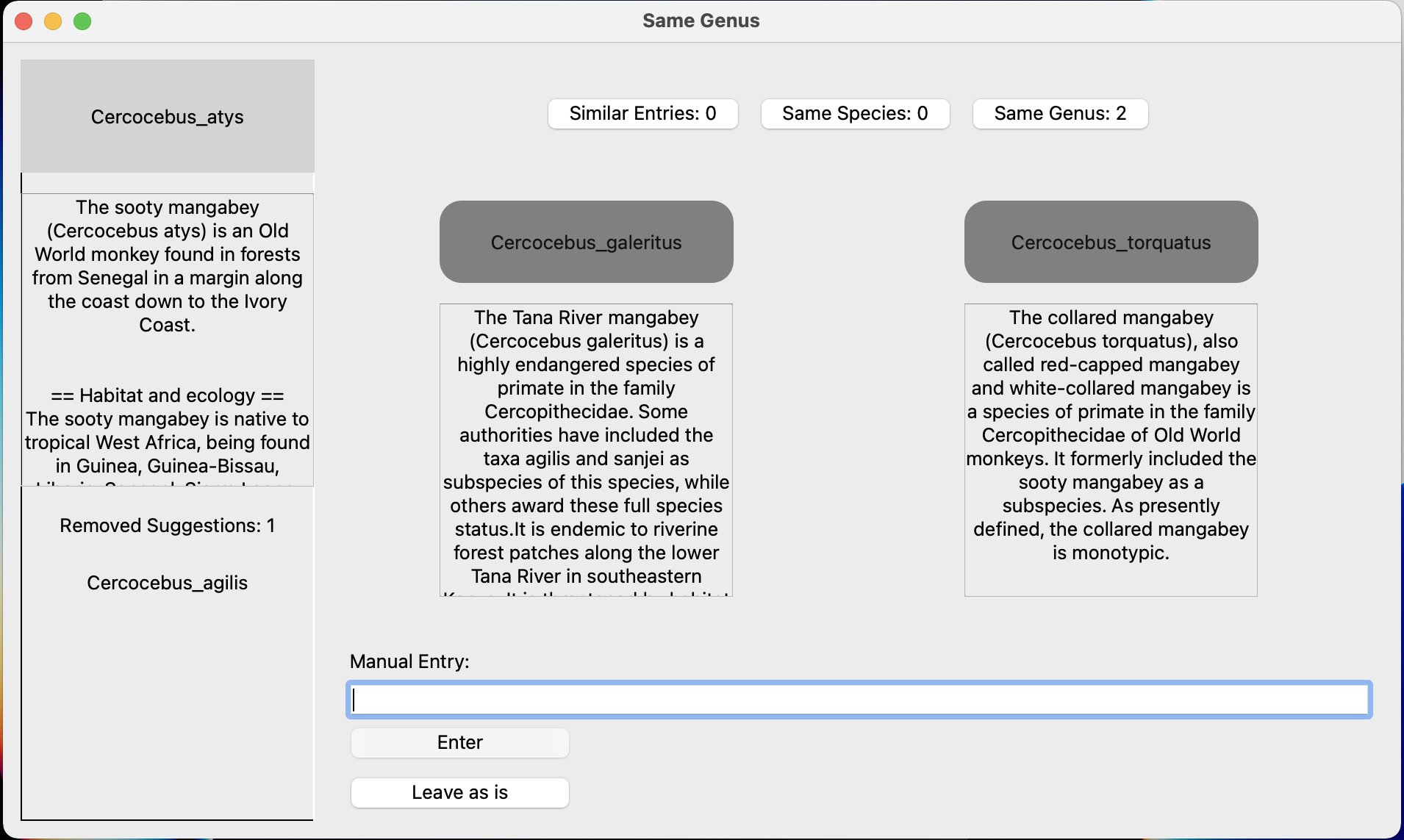
Here's an example where I might want to use the "Same Genus" option. This can be useful if you don't have many taxa in that genus and it doesn't really matter which species in the genus you use: Here I have data for Cercocebus atys but that taxon isn't in the tree. I could use a different Cercocebus species as a substitute. The 'removed suggestions' in the bottom left tells me that C. agilis is already matched between the data and tree. Since I know that C. atys and C. torquatus are sister species, both equally closely related to C. agilis, I can use C. torquatus instead.
Troubleshooting
'phylo-match' is not recognized as an internal or external command,
operable program or batch file.
This error usually means python has not been added to your PATH variable. See adding python to PATH tutorial or similar for details.
Contributing
Pull requests are welcome. For major changes, please open an issue first to discuss what you would like to change.
Please make sure to update tests as appropriate.
License
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file phylo_match-0.1.1.tar.gz.
File metadata
- Download URL: phylo_match-0.1.1.tar.gz
- Upload date:
- Size: 26.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.8.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d773062ac2ac42680a53dcaac9eca6205b07e7e6d0c161c4d4bee09a6e34058f
|
|
| MD5 |
b359995a4b0dfc05ffe2f32424f6fa97
|
|
| BLAKE2b-256 |
c7791a840be1097a8689296a4a79b708d3b6177f38eafd0d3c72f1c34f33f4d3
|
File details
Details for the file phylo_match-0.1.1-py3-none-any.whl.
File metadata
- Download URL: phylo_match-0.1.1-py3-none-any.whl
- Upload date:
- Size: 26.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.8.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c15f40f49dd9c07ff0c70624c5712d3c6e0609a8a1abb95323d1c4f005ed18a7
|
|
| MD5 |
93ca6c9cbe20257a23f43844a61fdb85
|
|
| BLAKE2b-256 |
de5772b72b7ae7f11fcf9b17b815b858e608bc73c5934f9fcea29adc9a3e7bfa
|







