phyloframe





Dataframe-based tools for working with phylogenetic trees.
- Free software: MIT license
- Documentation: https://phyloframe.readthedocs.io
- Repository: https://github.com/mmore500/phyloframe
Why a DataFrame-based Tree Representation?
The R ecosystem's success with the ape data structure demonstrates the utility of edge matrix tree representations --- phyloframe pushes this idea further with a fully tabular format hosted within DataFrame objects (e.g., pd.DataFrame, pl.LazyFrame, pl.DataFrame, etc.).
DataFrames are scripting-friendly and end-user extensible, enabling a composable, interoperable, high-performance ecosystem for phylogenetic analysis --- in applications to our work, scalable to billion-tip phylogenies.
Fast and highly portable load/save.
Use pandas.read_csv, polars.read_parquet, etc. --- libraries transparently fetch from URLs, cloud providers (S3, Google Cloud, etc.).
Fast, full-featured Newick format I/O support.
Benefit from modern tabular data formats. Granular deserialization of selected columns (e.g., Parquet), transparent compression configuration (e.g., Parquet), columnar compression for efficient storage, categorical strings, and explicit column typing.
Benefit from modern high-performance dataframe tooling. Lazy query optimization (e.g., Polars), larger-than-memory streaming operations (e.g., Polars), distributed computing operations (e.g., Dask), multithreaded operations (e.g., Polars), vectorized operations (e.g., NumPy), and just-in-time compilation (e.g., Numba).
Benefit from rich, expressive dataframe functionality. Leverage powerful querying and transformation APIs (e.g., Polars expressions, Pandas indexing), enabling flexible filtering, bulk column calculations, grouped aggregations, join/merge operations, and chained transformations directly over tree data without manual loops.
Cache-friendly, memory-efficient, flexible data structure. Data occupies contiguous arrays, expediting tree creation and topological order traversals (e.g., parents before children or vice versa). Base memory footprint is lightweight (e.g., as little as 32 bits per node), but can be dynamically augmented to expedite traversals and calculations (e.g., linked list over children via DataFrame columns for first child/next sibling indices).
Rich interoperative ecosystem.
Multi-language interoperation (e.g., possible future support for zero-copy interop between R and Python via reticulate and Arrow; possible future support for zero-copy Polars DataFrames shared between Rust and Python).
Multi-library interoperation (e.g., highly-optimized or zero-copy interoperation between Polars and Pandas; Python dataframe protocol).
Compatibility with existing alife data standards ecosystem.
Performance
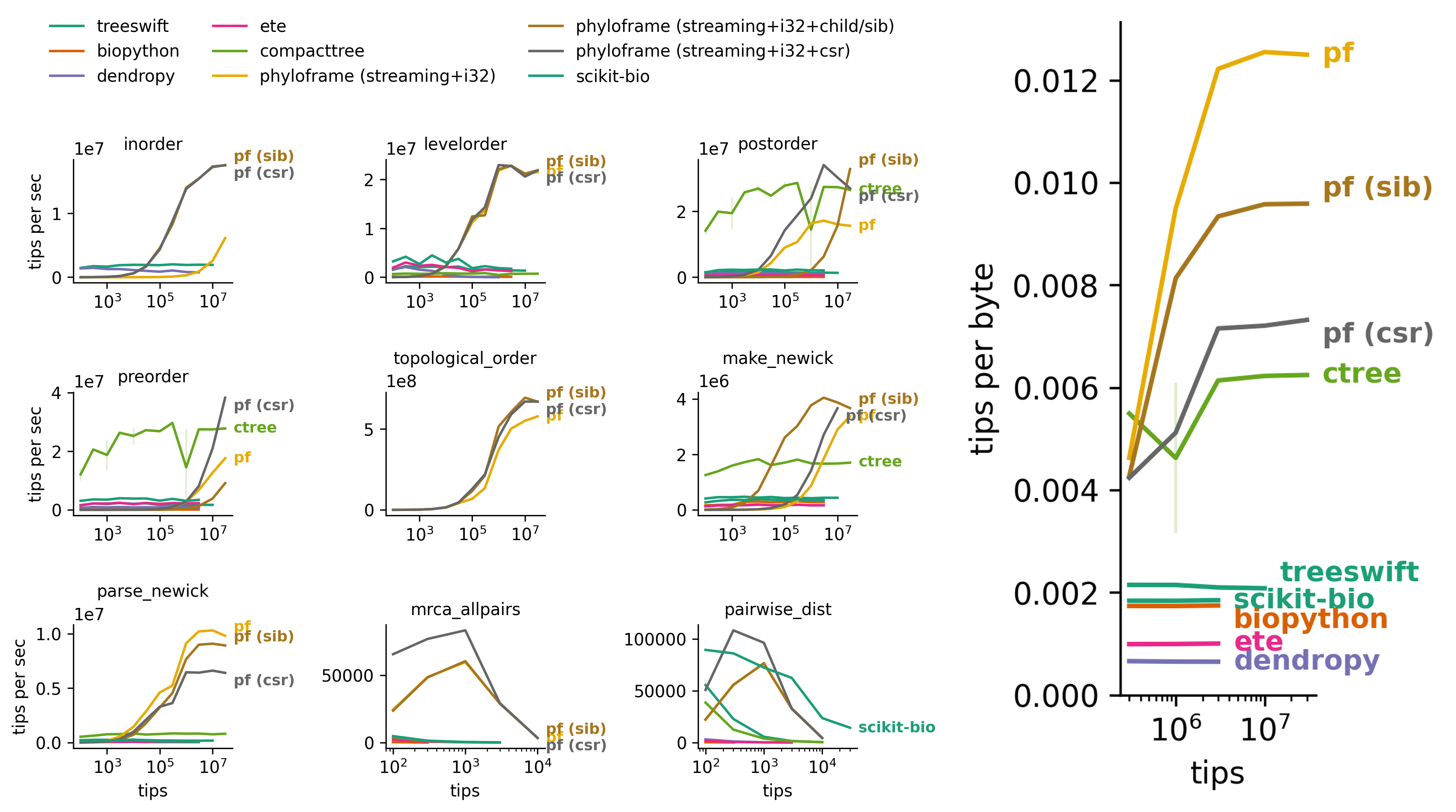
At large tree sizes, phyloframe improves speed and memory-efficiency for most operations.
Notably, newick reads and topological-order tree traversal (i.e., parents before children) are up to 10× faster than existing tools --- including implementations backed by native code. Newick writes are up to 2× faster.
Benchmarked operations include tree traversals, newick read/write, and pairwise operations.
Install
python3 -m pip install "phyloframe[jit]==0.10.1"
The [jit] extra installs Numba for just-in-time compilation, providing native-level performance for many operations.
JIT dependency is strongly recommended.
A containerized release of phyloframe is available via ghcr.io
singularity exec docker://ghcr.io/mmore500/phyloframe:v0.10.1 python3 -m phyloframe --help
Quickstart
Phyloframe represents phylogenies as DataFrames in the alife standard format.
from phyloframe import legacy as pfl
# Parse a Newick tree (already in working format)
df = pfl.alifestd_from_newick("((A:1,B:2):3,(C:4,D:5):6);")
# Mark properties and transform using df.pipe() (pandas syntactic sugar)
df = (
df.pipe(pfl.alifestd_mark_leaves)
.pipe(pfl.alifestd_mark_node_depth_asexual)
.pipe(pfl.alifestd_collapse_unifurcations)
)
print("leaf count:", pfl.alifestd_count_leaf_nodes(df))
print(df[["id", "ancestor_id", "is_leaf", "node_depth"]].head())
The legacy module (from phyloframe import legacy) provides all current phyloframe operations.
The legacy API is stable and will continue to be maintained for backward compatibility.
A redesigned API will accompany phyloframe v1.0.0.
For a deeper introduction covering tree representation semantics, tree creation, tree computations, tree transforms, Polars, CLI, JIT compilation, and more, see the full quickstart guide.
Citing
If phyloframe contributes to a scholarly work, please cite it as
Moreno M. A., Sukumaran J., Zaman L., & Dolson E. (2026). PhyloFrame: A DataFrame-based Library for Fast, Flexible Phylogenetic Computation. arXiv preprint arXiv:2605.28545. https://doi.org/10.48550/arXiv.2605.28545
@misc{moreno2026phyloframe,
doi={10.48550/arXiv.2605.28545},
url={https://arxiv.org/abs/2605.28545},
title={PhyloFrame: A DataFrame-based Library for Fast, Flexible Phylogenetic Computation},
author={Matthew Andres Moreno and Jeet Sukumaran and Luis Zaman and Emily Dolson},
year={2026},
eprint={2605.28545},
archivePrefix={arXiv},
primaryClass={q-bio.PE},
}
And don't forget to leave a star on GitHub!
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file phyloframe-0.10.1.tar.gz.
File metadata
- Download URL: phyloframe-0.10.1.tar.gz
- Upload date:
- Size: 2.0 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9d8adf4892f266f0c9dcb574a8abaa9b2126a32880b6cc6f14bc6922d45104ae
|
|
| MD5 |
fc28a1d8d0e86798f560742f060e7925
|
|
| BLAKE2b-256 |
bfa83a2ce49bfc8aa0e947837df81d967412f4c208393eac24385ec10ed92e15
|









