Analyze and manipulate phylogenetic trees

Project description
phylotreelib


phylotreelib is a Python library for reading, writing, analyzing, manipulating, comparing, and summarizing phylogenetic trees, and is designed to be used in scripts or interactively in notebooks.
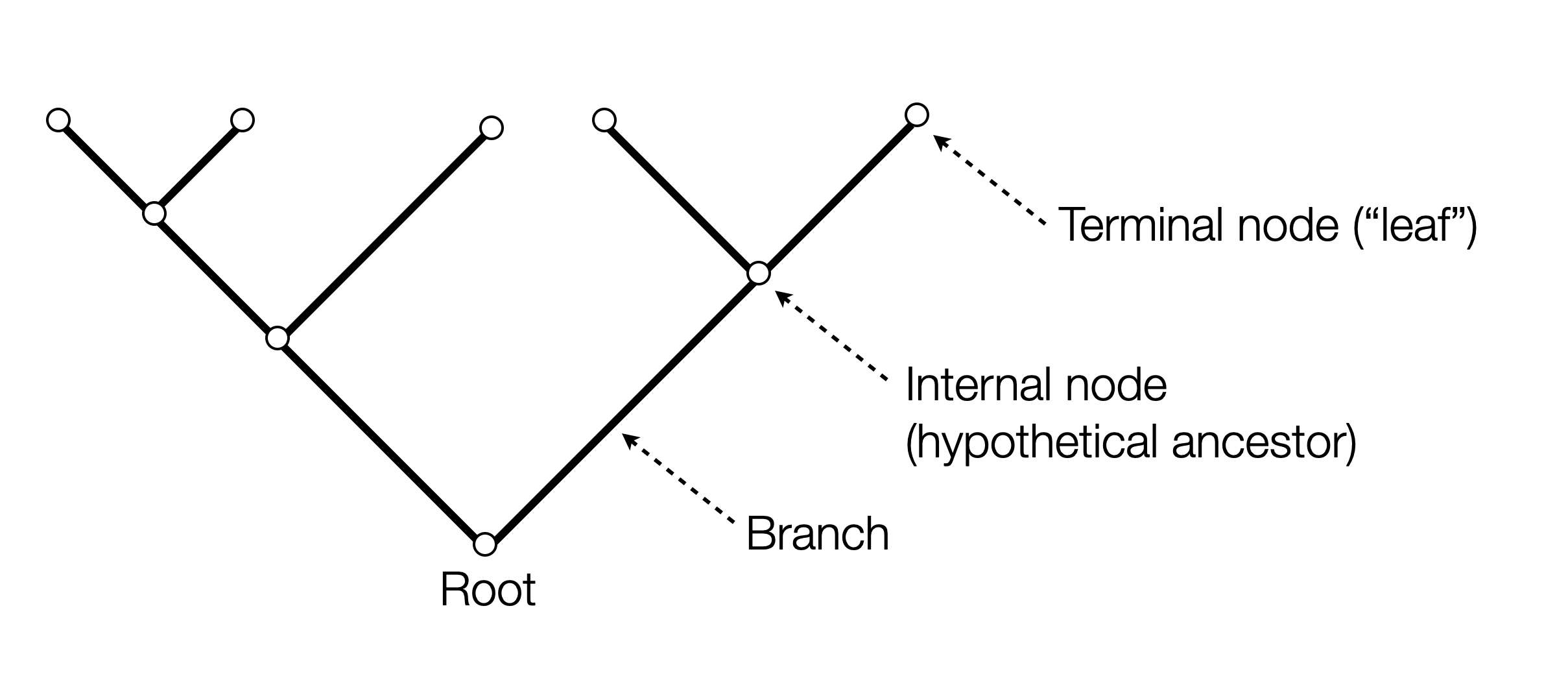
Main capabilities include:
- reading and writing Newick and NEXUS tree files
- querying tree structure (MRCA, descendants, paths, distances, neighborhoods)
- rerooting by midpoint, outgroup, or minimum-variance criteria
- pruning leaves or subtrees, regrafting, and other topology edits
- computing tree distances such as Robinson–Foulds and path-difference distance
- building trees from distance matrices (neighbor joining and UPGMA)
- summarizing posterior tree samples into consensus, MCC, MBC, and HIPSTR-style summary trees
- exporting trees with labels, metadata, and optional coloring for visualization in tree viewers (e.g., FigTree)
- library has been optimized for high speed and low memory consumption
- NOTE: labels are interpreted as belonging to branches (bipartitions), not to internal nodes, and this association is maintained after re-rooting etc.
Version 2.0.0
Version 2 has recently been released, and contains a number of changes to the API compared to version 1.x.x.
- See the Quick start below for updated examples.
- For details on API changes and how to update existing scripts, see Upgrading from 1.x to 2.x.
- For a feature overview, see Changelog (2.0.0).
Installation
python3 -m pip install phylotreelib
Upgrade to the latest version:
python3 -m pip install --upgrade phylotreelib
Documentation
This repository uses three main documentation files:
README.md— (this file) overview and quick startdocs/recipes.md— copy/paste-ready examples and workflowsdocs/api.md— more extensive reference to the API, plus advanced notes
The README gives examples of the most common workflows. More specialized details, and the theory behind some of the summary-tree machinery, are in the docs/ files.
Quick start
Terminology note: tree orientation and "height" vs "depth"
Note for users upgrading from version 1: In phylotreelib v. 2, trees are now treated as having the root at the top and leaves at the bottom — matching the standard convention in most phylogeny software and computer science.
Accordingly, it uses:
- height(node) = distance from the tips (leaves) to the node (i.e., "time before most recent leaf")
In v1.x, this concept was called depth. All depth-based names were renamed to height-based names in v2.0.0. See Upgrading from 1.x to 2.x for the full rename table.
Construct a tree from a Newick string, print tabular representation
import phylotreelib as pt
tree = pt.Tree.from_string("(Orangutan:4,(Gorilla:3,(Human:2,(Chimpanzee:1,Bonobo:1):1):1):1);")
print(tree)
Output:
+---------------------------------------+
| parent | child | branchlen |
+---------------------------------------+
| 0 | 1 | 1 |
| 0 | Orangutan | 4 |
| 1 | 2 | 1 |
| 1 | Gorilla | 3 |
| 2 | 3 | 1 |
| 2 | Human | 2 |
| 3 | Bonobo | 1 |
| 3 | Chimpanzee | 1 |
+---------------------------------------+
Tree length: 14.0
5 Leaves:
----------
Bonobo
Chimpanzee
Gorilla
Human
Orangutan
Read one tree from file, reroot it, print all root-to-tip distances
import phylotreelib as pt
with pt.Nexustreefile("mytreefile.nexus") as treefile:
tree = treefile.readtree()
tree.rootminvar()
for tip in sorted(tree.leaves):
dist = tree.nodedist(tree.root, tip)
print(f"{tip:<12s} {dist:.3f}")
Query tree structure
The examples below illustrate some of the most useful tree queries: finding an MRCA, listing descendants, computing patristic distance, finding nearby leaves by patristic distance.
import phylotreelib as pt
tree = pt.Tree.from_string("(Orangutan:4,(Gorilla:3,(Human:2,(Chimpanzee:1,Bonobo:1):1):1):1);")
mrca = tree.find_mrca({"Human", "Chimpanzee"})
print("MRCA:", mrca)
print("Descendant leaves:", tree.remote_children(mrca))
print("Distance Human–Bonobo:", tree.nodedist("Human", "Bonobo"))
print("Leaves within distance 4.5 of Human:", tree.nearleafs("Human", 4.5))
print("Nearest 3 leaves to Human:", tree.nearest_n_leaves("Human", 3))
Remove selected leaves, keep rest of topology
import phylotreelib as pt
tree = pt.Tree.from_string("((A:1,B:1):1,((C:1,D:1):1,(E:1,F:1):1):1);")
tree.remove_leaves(["B", "D"])
print(tree.newick())
Output:
(((E:1,F:1):1,C:2):1,A:2);
Perform a specified SPR move
import phylotreelib as pt
tree = pt.Tree.from_string("(((A:1,B:1):1,(C:1,D:1):1):1,(E:1,F:1):1);")
prune_node = tree.find_mrca({"C", "D"})
regraft_node = tree.find_mrca({"E", "F"})
tree.spr(prune_node=prune_node, regraft_node=regraft_node)
print(tree.newick())
Perform a semi-random or fully random SPR move
import phylotreelib as pt
tree = pt.Tree.randtree(ntips=12, randomlen=True)
print(tree.newick(printdist=False))
# semi-random: choose prune node, randomize regraft point
prune_node = next(iter(tree.possible_spr_prune_nodes()))
tree.spr(prune_node=prune_node)
print(tree.newick(printdist=False))
# fully random
for _ in range(10):
tree.spr()
print(tree.newick(printdist=False))
Summarize posterior tree samples
If you are working with posterior tree samples (e.g. from BEAST/MrBayes), you can build a summary tree in a few explicit steps:
- TreeSummary accumulates frequencies and (optionally) branch-length/node-height statistics
- SummaryTreeBuilder constructs a summary-tree topology
- TreePostProcessor adds attributes to the tree annotating it with e.g. support values / branch-length stats
- PrintSpec controls what attributes are written as branch labels and/or Nexus meta-comments
For command-line use, the separate tool sumt wraps these steps in a single command.
# Download an example tree-sample file (run these 3 lines only once)
import urllib.request
URL = "https://raw.githubusercontent.com/agormp/phylotreelib/main/tests/primate-mtDNA.trees"
urllib.request.urlretrieve(URL, "primate-mtDNA.trees")
import phylotreelib as pt
# 1) Accumulate bipartition frequencies + mean branch lengths
ts = pt.TreeSummary(trackbips=True, trackblen=True)
with pt.Nexustreefile("primate-mtDNA.trees") as tf:
for tree in tf:
ts.add_tree(tree)
# 2) Build a majority-rule consensus topology
stb = pt.SummaryTreeBuilder(ts)
sumtree = stb.contree(cutoff=0.5, allcompat=False)
# 3) Annotate/paint the tree from the collected summaries
tpp = pt.TreePostProcessor(ts)
tpp.annotate_sumtree(sumtree)
# 4) Configure printing (labels + optional meta-comments)
pt.configure_sumtree_printing(sumtree, treetype="con", blen="biplen", print_meta=True, precision=7)
print(sumtree.newick())
Compute and export a summary tree in one step
# Download an example tree-sample file (run these 3 lines only once)
import urllib.request
URL = "https://raw.githubusercontent.com/agormp/phylotreelib/main/tests/primate-mtDNA.trees"
urllib.request.urlretrieve(URL, "primate-mtDNA.trees")
import phylotreelib as pt
ts = pt.TreeSummary(
trackbips=True,
trackclades=True,
trackroot=True,
trackblen=True,
trackheight=True,
trackrootblen=True,
tracktopo=True,
trackci=True,
)
with pt.Nexustreefile("primate-mtDNA.trees") as tf:
for tree in tf:
ts.add_tree(tree)
sumtree = pt.build_sumtree(ts, treetype="mcc", blen="cladeheight", rooting=None,)
pt.configure_sumtree_printing(
sumtree,
treetype="mcc",
blen="cladeheight",
trackci=True,
print_meta=True
)
with open("posterior_summary.nexus", "w") as out:
out.write(sumtree.nexus())
Save in different formats and include metadata
The printing helpers let you choose labels, branch lengths, and metadata comments independently of the tree itself.
import phylotreelib as pt
tree = pt.Tree.from_string("((A:1,B:1):1,(C:1,D:1):1);")
# attach a branch attribute that can later be printed
mrca = tree.find_mrca({"A", "B"})
parent = tree.parent(mrca)
tree.set_branch_attribute(parent, mrca, "posterior", 0.97)
pt.configure_basic_printing(
tree,
print_meta=True,
branch_attrs=("posterior",),
precision=6,
)
print(tree.newick())
print(tree.nexus(translateblock=True))
Color a selected set of leaves in NEXUS output
This is useful for visualization in for instance FigTree. Here, the leaves to color are obtained by a biological query on the tree (specifically: the descendants of the MRCA of the mink leaves).
import phylotreelib as pt
tree = pt.Tree.from_string("((Human_A:1,Human_B:1):1,(Mink_A:1,Mink_B:1):1,(Bat_A:1,Bat_B:1):1);")
mink_mrca = tree.find_mrca({"Mink_A", "Mink_B"})
color_me = sorted(tree.remote_children(mink_mrca))
print(tree.nexus(translateblock=False, colorlist=color_me,
colorfg="#FF0000", colorbg="#000000"))
Core concepts
Tree class
Tree is the central class. Most workflows involve reading or constructing one or more Tree objects and then querying or modifying them.
Useful attributes:
tree.leaves— set of leaf namestree.intnodes— set of internal node IDstree.nodes— set of all nodestree.root— current root node ID
A tabular representation can be obtained with:
print(tree)
Tree files
Recommended usage:
import phylotreelib as pt
with pt.Treefile("trees.nex") as tf:
tree = tf.readtree()
Available classes:
Treefile— file-format autodetectionNewicktreefileNexustreefile
Tree sets and tree summaries
TreeSetstores multiple trees with the same leaves.TreeSummaryaccumulates summary statistics across trees.SummaryTreeBuilderconstructs summary trees such as consensus, MCC, MBC, and HIPSTR.TreePostProcessoradds attributes to the tree annotating it with e.g. support values / branch-length stats.
Where to go next
- For more worked examples, see
docs/recipes.md - For the API reference and advanced notes, see
docs/api.md
Related tools
sumt— command-line interface for summary-tree workflows built onphylotreelib
Changelog (2.0.0)
Added
- Explicit summary-tree pipeline via
SummaryTreeBuilderandTreePostProcessor(with optionalCAHeightEstimatorfor CA-heights and credible intervals). - Credible interval support via
QuantileAccumulatorandtrackci/ci_probsinTreeSummary. - PrintSpec plus helpers
configure_basic_printing(...)andconfigure_sumtree_printing(...)for consistent Newick/NEXUS output.
Changed
Tree.graft(...)now grafts onto a specified edge(parent -> child)and usesconnect_length/connect_branchstructfor the connecting branch.Tree.spr(...)updated to match the new grafting model and (by default) preserve total tree length.Tree.subtree(...)/Tree.prune_subtree(...)now return(subtree, basal_branch); leaf subtrees return the leaf name asstr.Tree.reroot(...)signature updated to use the same distance/fraction placement convention as other editing methods.Tree.newick(...)/Tree.nexus(...)now treatNoneas "use PrintSpec defaults".
Renamed (terminology: depth → height)
All names relating to node heights — the distance from the tips to an internal node, i.e. "time before the most recent leaf" — have been renamed from depth-based to height-based. This aligns with the convention used in most phylogenetic software and programming libraries. This is a breaking change; the old names are no longer available.
| v1.x name | v2.x name |
|---|---|
TreeSummary(..., trackdepth=True, ...) |
TreeSummary(..., trackheight=True, ...) |
Tree.nodedepth(node) |
Tree.nodeheight(node) |
Tree.set_blens_from_depths() |
Tree.set_blens_from_heights() |
TreePostProcessor.set_mean_node_depths(tree) |
TreePostProcessor.set_mean_node_heights(tree) |
CADepthEstimator |
CAHeightEstimator |
build_sumtree(..., blen="meandepth", ...) |
build_sumtree(..., blen="cladeheight", ...) |
build_sumtree(..., blen="cadepth", ...) |
build_sumtree(..., blen="caheight", ...) |
Node attribute depth |
Node attribute height |
Node attribute depth_sd |
Node attribute height_sd |
Node attribute depth_median |
Node attribute height_median |
Deprecated
Tree.deroot()→ useTree.collapse_bifurcating_root().
Fixed / improved
- Multiple refactorings leading to performance improvements (speed and memory) and bug fixes.
Upgrading from 1.x to 2.x
phylotreelib 2.0 introduces a few intentional API breaks to make tree editing, summary trees, and printing more explicit and less "magic".
If you maintain scripts that call phylotreelib directly, the sections below show the key signature/return changes and how to update call sites.
Terminology rename: depth → height
In v1.x, the distance from the tips to an internal node was called depth. In v2.0.0 this has been renamed to height throughout the library. The rename is purely mechanical: a search-and-replace across your scripts using the table in the Changelog above is sufficient.
The affected call sites are:
TreeSummaryconstructor:trackdepth=→trackheight=build_sumtreeandconfigure_sumtree_printing:blen="cladedepth"→"cladeheight",blen="cadepth"→"caheight"Treemethods:nodedepth()→nodeheight(),set_blens_from_depths()→set_blens_from_heights()- Node attributes on summary trees:
depth/depth_sd/depth_median→height/height_sd/height_median
Summary trees
TreeSummary.compute_sumtree(...) is still available as a convenience wrapper (so many 1.x workflows continue to work), but the preferred style is now the explicit pipeline:
TreeSummary-->SummaryTreeBuilder-->TreePostProcessor(+ optionalCAHeightEstimator) -->PrintSpec
See the "Summarize posterior tree samples" quick-start example above, and the worked examples in docs/recipes.md.
Tree.graft: signature change
Old (1.x):
Tree.graft(self, other, node1, node2=None, blen1=0, blen2=0,
graftlabel=None, graft_with_other_root=False)
Meaning (1.x): attach other on incoming branch to node1 (possibly re-rooting other around node2): insert a new internal node on the incoming branch to node1 with length blen1, and connect other below that (meaning closer to the tips) with length blen2 (or optionally skip the extra basal branch).
New (2.x):
Tree.graft(self, other, parent, child,
dist_from_parent=None, frac_from_parent=None,
connect_length=0.0, connect_branchstruct=None, graftlabel=None)
Meaning (2.x): insert the graft point on an existing edge (parent -> child) and then attach other (or a single leaf name) below that graft point via a separate connecting branch.
How to convert typical 1.x calls
- Grafting on incoming edge to node1 (typical old usage)
Old:
tree.graft(other, node1=X, blen1=blen1, blen2=blen2)
New (equivalent intent):
parent = tree.parent(X)
tree.graft(
other,
parent=parent, child=X,
# place graftpoint on the incoming edge to X:
dist_from_parent=blen1, # if you want a specific distance
# or frac_from_parent=0.5, # if you just want "midway"
connect_length=blen2,
)
- Old
graft_with_other_root=True
In 2.x, the caller should explicitly decide what other is before calling graft(...):
- If you want to attach a single leaf, pass a string
other="TaxonName". - If you want to attach a Tree with a specific root, re-root
otherfirst (e.g.other.reroot(...)) and then callgraft(...).
Tree.spr: signature change (and length preservation)
Old (1.x):
Tree.spr(self, prune_node=None, regraft_node=None)
New (2.x):
Tree.spr(self, prune_node=None, regraft_node=None,
dist_from_parent=None, frac_from_parent=None)
New behaviour highlights:
- Regrafting happens by inserting a graft point on the incoming edge to
regraft_node. - If
dist_from_parentandfrac_from_parentare bothNone, the placement is randomized (uniform frac in (0,1)). - Total tree length is preserved by default (the regraft edge is split according to
dist_from_parentorfrac_from_parent; the length of the incoming basal branch to the prune_node is reused as the connecting branch length).
How to convert
Old:
tree.spr(prune_node=P, regraft_node=R)
New (same defaults):
tree.spr(prune_node=P, regraft_node=R)
New (explicit placement on incoming edge to regraft_node):
# parent = tree.parent(R)
tree.spr(prune_node=P, regraft_node=R, frac_from_parent=0.25)
# (places the graftpoint 25% down the edge parent->R)
Tree.subtree / Tree.prune_subtree: return value change
Old (1.x):
Tree.subtree(self, basenode, return_basalbranch=False) -> Tree
Tree.prune_subtree(self, basenode) -> Tree
New (2.x):
Tree.subtree(self, basenode) -> (subtree, basal_branch)
Tree.prune_subtree(self, basenode) -> (subtree, basal_branch)
Notes (2.x):
basal_branchis a copy of the Branchstruct on the incoming edge tobasenode.- If
basenodeis a leaf:subtreeis the leaf name (str), not a mini tree.
How to convert
Old:
sub = tree.subtree(node)
New:
sub, basal = tree.subtree(node)
Old:
sub = tree.prune_subtree(node)
New:
sub, basal = tree.prune_subtree(node)
# basal.length is the length removed when detaching the subtree
Tree.reroot: signature change
Old (1.x):
Tree.reroot(self, node1, node2=None, polytomy=False, node1dist=0.0)
New (2.x):
Tree.reroot(self, node1, node2=None, polytomy=False,
dist_from_node1=None, frac_from_node1=0.5)
Conversion:
- If you previously used
node1dist, rename it todist_from_node1. - If you previously relied on the default placement, you can now do that via
frac_from_node1(default 0.5).
Root-collapsing helper rename
Old (1.x):
Tree.deroot(self)
New (2.x):
Tree.collapse_bifurcating_root(self)
(And Tree.deroot() is kept as a deprecated alias.)
Printing: PrintSpec defaults
Old (1.x):
Tree.newick(self, printdist=True, printlabels=True, labelfield='label', precision=6, ...)
Tree.nexus(self, printdist=True, printlabels=True, labelfield='label', precision=6, ...)
New (2.x):
Tree.newick(self, printdist=None, printlabels=None, labelfield=None, precision=None, ..., print_meta=None)
Tree.nexus(self, printdist=None, printlabels=None, labelfield=None, precision=None, ..., print_meta=None)
In 2.x, passing None means "use the Tree's current PrintSpec default". This makes it easy to configure printing once:
pt.configure_basic_printing(tree, precision=7, print_meta=True, branch_attrs=("posterior",))
print(tree.newick()) # uses PrintSpec
print(tree.nexus()) # uses PrintSpec
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file phylotreelib-2.3.0.tar.gz.
File metadata
- Download URL: phylotreelib-2.3.0.tar.gz
- Upload date:
- Size: 119.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.4
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
6c60fb21b3b641d05340ddd2e9b65d13d17d7998dd945b74e843e3b486a7f937
|
|
| MD5 |
d304ee9f49d5649cad26528efb5133e8
|
|
| BLAKE2b-256 |
2e8a853027539ad2532800e94e07d230e2e6baff75211ac4d51a7c97c7a05be8
|
File details
Details for the file phylotreelib-2.3.0-py3-none-any.whl.
File metadata
- Download URL: phylotreelib-2.3.0-py3-none-any.whl
- Upload date:
- Size: 97.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.4
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
899cfd823d79e36ec94e9e434d7b19dbd4b6cdfb4b06b282abaaef59183da62c
|
|
| MD5 |
13332734e26e4c376942396b6f162465
|
|
| BLAKE2b-256 |
427621fb286e1ee57e9fa98fa4ddbbe1bd5b8280a7de89a70c8bed16ed9d17a1
|







