Tool for the integration of viral consensus sequences obtained by de novo and mapping strategies, supported by prior information.

Project description
PriorCons
This repository provides tools to:
-
Generate Integrated Consensus (
integrate_consensus.py)
Produces a high-quality viral consensus by strategically using ABACAS sequences to fill missing regions in the mapping consensus. It employs a sliding-window approach that verifies the evolutionary plausibility of ABACAS content against empirical priors before incorporation. -
Build Evolutionary Priors (
build_priors.py)
Constructs empirical prior distributions from large multiple-sequence alignments. These priors model expected genetic variation across genomic windows and provide likelihood thresholds for quality control during consensus integration. -
Access Supporting Utilities (
utilsscripts)
Provides modular helper functions for alignment processing, window scoring, and consensus construction used by both main workflows.
Installation
pip install priorcons
CLI usage
# Create priors
priorcons build-priors --input sequences.fasta --ref REF_ID --output priors.parquet
# Run consensus integration
priorcons integrate-consensus --input alignment.aln --ref REF_ID --prior priors.parquet --output_dir results
🚀 Main Script: integrate_consensus.py
This is the entrypoint of the tool. It creates a integrated consensus sequence by combining mapping consensus and ABACAS output, both aligned to a reference sequence, but only after performing quality control (QC) at the window level.
🔑 Inputs
--input→ path to an alignment file (.aln) containing at least:- 1º Reference sequence
- 2º Mapping consensus sequence
- 3º ABACAS consensus sequence
The sequences in the alignment file must be provided in the specified order, as they will be identified by their position.
-
--ref→ ID of the reference sequence in the alignment. -
--prior→ path to a priors table (.parquet) generated withbuild_priors.py. -
--output_dir→ directory to save the results.
🧪 Workflow
- Start with mapping consensus as the baseline
- Identify missing/unreliable regions in mapping consensus
- For each window:
- If mapping has coverage → keep mapping sequence
- If mapping has missing data → evaluate ABACAS for that window:
- Check fragmentation and quality
- Verify evolutionary plausibility using priors (nLL score)
- If ABACAS passes QC → use ABACAS to fill missing regions
- Construct final consensus combining mapping baseline with validated ABACAS fills
- Restore mapping-specific insertions
- QC reporting: compute coverage, substitutions, and insertion metrics comparing the final integrated consensus to MAPPING.
📦 Outputs
The script produces three files inside --output_dir:
-
Integrated consensus FASTA
- File:
<basename>-INTEGRATED.fasta - Contains the final consensus sequence after merging and reinserting insertions.
- File:
-
Window QC trace (CSV)
- File:
windows_trace.csv - One row per window, recording:
start,end→ genomic coordinates.MISSING_MAPPING,MISSING_ABACAS→ counts of missing bases.ABACAS_MORE_INFO→ whether ABACAS has fewer missing bases than MAPPING.ABACAS_FRAGMENTS→ fragmentation level of ABACAS in this window (keep: 0 < n fragments < 3 ).WINDOW_PRIOR_nLL_p95→ threshold from priors.WINDOW_SCORE_nLL→ score of ABACAS in this window.WINDOW_QC_PASSED→ True/False decision.
- File:
-
Consensus QC summary (JSON)
- File:
qc.json - Provides overall metrics comparing the MAPPING consensus and the integrated consensus:
MAPPING_COVERAGE→ % of genome covered in MAPPING.FINAL_COVERAGE→ % of genome covered in integrated consensus.MAPPING_SUBSTITUTIONS→ substitutions vs. reference in MAPPING.FINAL_SUBSTITUTIONS→ substitutions vs. reference in integrated consensus.EXPECTED_SUBSTITUTIONS→ expected number of substitutions, extrapolated from mapping.OBS-EXP_SUBSTITUTIONS→ difference between observed and expected substitutions.N_INSERTIONS→ number of insertions added back.TOTAL_INSERTIONS_LENGTH→ total inserted length.INSERTIONS→ list of insertions with their coordinates.
- File:
▶️ Example run
python integrate_consensus.py \
--input /path/to/<sample_name>.aln \
--ref RSV_BD \
--prior /path/to/RSVBD_win100_ovlp50_priors.parquet \
--output_dir results
This will generate:
results/<sample_name>-INTEGRATED.fastaresults/windows_trace.csvresults/qc.json
🛠 Script: build_priors.py
This script creates empirical priors (overlapped windows) from a large multiple sequence alignment.
These priors are later used by integrate_consensus.py to evaluate windows.
🔑 Inputs
-i / --input→ aligned FASTA file with multiple sequences.-r / --ref→ ID of the reference sequence.-o / --output→ output file (.parquet).--win→ window size (default: 100).--overlap→ overlap size (default: 10).
▶️ Example run
python build_priors.py \
-i alignment.fasta \
-r ReferenceID \
-o priors.parquet \
--win 100 \
--overlap 10
📦 Output
A .parquet file with one row per window, containing:
start,end→ window coordinates.nLL_p95,nLL_p99→ empirical thresholds.profile→ base probability distributions for each position in the window.
🧮 Methodology (build_priors.py)
1. Probability distributions per position
For each window of size W bases (e.g., W = 100), and for each position j within that window, we compute the probability of observing each nucleotide:
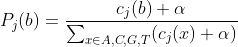
Where:
= number of sequences with base
at position
.
= pseudocount (Laplace smoothing, default
) to avoid zero probabilities.
- Bases
Nare ignored in the counts.
This gives a per-position categorical distribution.
2. Log-likelihood of a sequence in a window
Given a query sequence 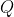
For each valid (non-N) position 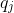
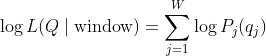
The normalized negative log-likelihood (nLL) is:
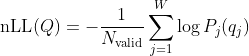
Where:
= number of positions in the window where
Nbase.
Smaller nLL values indicate sequences more likely under the empirical profile.
3. Empirical priors
To characterize "normal variation" for each window:
- Score all sequences from the alignment against the window profile.
- Collect the distribution of nLL values.
- Extract percentiles (e.g., 95th and 99th) to serve as thresholds.
Thus, for each window we store:
- The distribution (profile).
- Empirical thresholds:
nLL_p95andnLL_p99.
A new sequence can later be compared:
- If
nLL < nLL_p95→ typical. - If
nLL > nLL_p99→ unusually variable, possibly unreliable region.
� Supporting utils
Several utility scripts provide reusable functions for both processes:
-
utils.py → basic alignment and scoring functions:
load_alignment,extract_ref_positions,sliding_windows,score_window.
-
utils_integrate_consensus.py → additional helpers for consensus integration:
- missingness and fragmentation counts,
- insertion handling,
- QC calculations,
- consensus merging,
- window evaluation wrapper.
These modular functions keep the pipeline clean and reusable.
QC ANALYSIS
This tool allows also an analyis QC.
The input is a directory with all the results folders (one for each sample) there are stored the qc files:
- input: priorcons_path / / qc_files
It also need a gtf file and an outdir.
priorcons qc --input_dir /path/to/results/PRIORCONS/ \
--gff_file /path/to/rsv.gff \
--output_dir /path/to/output_dir_plots
Test dataset for integration in nf-core/viralrecon pipeline
https://zenodo.org/records/17454552/files/PriorCons_Test_data.zip?download=1


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file priorcons-0.1.1.tar.gz.
File metadata
- Download URL: priorcons-0.1.1.tar.gz
- Upload date:
- Size: 27.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8581286689aa85105f4a42c4d62cbd76d279099790d09316ad53cda131e603ed
|
|
| MD5 |
b469ded23103d9a33f33359f551c3981
|
|
| BLAKE2b-256 |
c23f65ebf49760c99f1f163ca14dea687d1ea0f28a75df097a919cf5cc8a1608
|
File details
Details for the file priorcons-0.1.1-py3-none-any.whl.
File metadata
- Download URL: priorcons-0.1.1-py3-none-any.whl
- Upload date:
- Size: 27.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
705630db6507000a26df7cb567e6590e7eb9d37ebe8925a05c51826c3959193d
|
|
| MD5 |
6f9ef3efc32daed59cba7a43c2973158
|
|
| BLAKE2b-256 |
243294bcb4fd9316c72abc431498680df068843b0d50ae0bf623cf62fd62ee37
|







