a slightly cleaned up installable version of ProteinMPNN

Project description
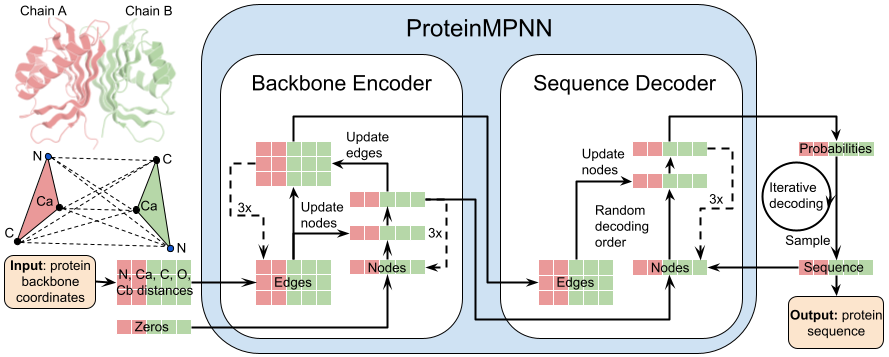
ProteinMPNN
"pip install proteinmpnn"
Requires Python >=3.9, <3.13
# Example usage
proteinmpnn --out-folder outdir --pdb-path foo.pdb
proteinmpnn --out-folder outdir --pdb-path foo.pdb --use-soluble-model
proteinmpnn --out-folder outdir --jsonl_path bar.jsonl
See original repo https://github.com/dauparas/ProteinMPNN for more examples and code for retraining.
Helper scripts: helper_scripts - helper functions to parse PDBs, assign which chains to design, which residues to fix, adding AA bias, tying residues etc.
Code organization:
protein_mpnn_run.py- the main script to initialialize and run the model.protein_mpnn_utils.py- utility functions for the main script.
Input flags for protein_mpnn_run.py:
argparser.add_argument("--suppress_print", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--ca_only", action="store_true", default=False, help="Parse CA-only structures and use CA-only models (default: false)")
argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;")
argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise")
argparser.add_argument("--use_soluble_model", action="store_true", default=False, help="Flag to load ProteinMPNN weights trained on soluble proteins only.")
argparser.add_argument("--seed", type=int, default=0, help="If set to 0 then a random seed will be picked;")
argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-log_prob to npy files")
argparser.add_argument("--path_to_fasta", type=str, default="", help="score provided input sequence in a fasta format; e.g. GGGGGG/PPPPS/WWW for chains A, B, C sorted alphabetically and separated by /")
argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position")
argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs")
argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)")
argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)")
argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output unconditional probabilities p(s_i given backbone) in one forward pass")
argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms")
argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target")
argparser.add_argument("--batch_size", type=int, default=1, help="Batch size; can set higher for titan, quadro GPUs, reduce this if running out of GPU memory")
argparser.add_argument("--max_length", type=int, default=200000, help="Max sequence length")
argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.2 0.25 0.5. Sampling temperature for amino acids. Suggested values 0.1, 0.15, 0.2, 0.25, 0.3. Higher values will lead to more diversity.")
argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/")
argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed")
argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB ")
argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed pdb into jsonl")
argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.")
argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions")
argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.")
argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias if neededi, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.")
argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.")
argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices")
argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm")
argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions")
argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs")
argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions")
Output example:
>3HTN, score=1.1705, global_score=1.2045, fixed_chains=['B'], designed_chains=['A', 'C'], model_name=v_48_020, git_hash=015ff820b9b5741ead6ba6795258f35a9c15e94b, seed=37
NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER/NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER
>T=0.1, sample=1, score=0.7291, global_score=0.9330, seq_recovery=0.5736
NMYSYKKIGNKYIVSINNHTEIVKALKKFCEEKNIKSGSVNGIGSIGSVTLKFYNLETKEEELKTFNANFEISNLTGFISMHDNKVFLDLHITIGDENFSALAGHLVSAVVNGTCELIVEDFNELVSTKYNEELGLWLLDFEK/NMYSYKKIGNKYIVSINNHTDIVTAIKKFCEDKKIKSGTINGIGQVKEVTLEFRNFETGEKEEKTFKKQFTISNLTGFISTKDGKVFLDLHITFGDENFSALAGHLISAIVDGKCELIIEDYNEEINVKYNEELGLYLLDFNK
>T=0.1, sample=2, score=0.7414, global_score=0.9355, seq_recovery=0.6075
NMYKYKKIGNKYIVSINNHTEIVKAIKEFCKEKNIKSGTINGIGQVGKVTLRFYNPETKEYTEKTFNDNFEISNLTGFISTYKNEVFLHLHITFGKSDFSALAGHLLSAIVNGICELIVEDFKENLSMKYDEKTGLYLLDFEK/NMYKYKKIGNKYVVSINNHTEIVEALKAFCEDKKIKSGTVNGIGQVSKVTLKFFNIETKESKEKTFNKNFEISNLTGFISEINGEVFLHLHITIGDENFSALAGHLLSAVVNGEAILIVEDYKEKVNRKYNEELGLNLLDFNL
score- average over residues that were designed negative log probability of sampled amino acidsglobal score- average over all residues in all chains negative log probability of sampled/fixed amino acidsfixed_chains- chains that were not designed (fixed)designed_chains- chains that were redesignedmodel_name/CA_model_name- model name that was used to generate results, e.g.v_48_020git_hash- github version that was used to generate outputsseed- random seedT=0.1- temperature equal to 0.1 was used to sample sequencessample- sequence sample number 1, 2, 3...etc
@article{dauparas2022robust,
title={Robust deep learning--based protein sequence design using ProteinMPNN},
author={Dauparas, Justas and Anishchenko, Ivan and Bennett, Nathaniel and Bai, Hua and Ragotte, Robert J and Milles, Lukas F and Wicky, Basile IM and Courbet, Alexis and de Haas, Rob J and Bethel, Neville and others},
journal={Science},
volume={378},
number={6615},
pages={49--56},
year={2022},
publisher={American Association for the Advancement of Science}
}
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file proteinmpnn-0.1.3.tar.gz.
File metadata
- Download URL: proteinmpnn-0.1.3.tar.gz
- Upload date:
- Size: 68.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.0.1 CPython/3.12.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c7b4a833cc63295f5ce99528efb96fcd372988d32db2650079d8d3a19a8adec1
|
|
| MD5 |
ef11a97d14c1aa388254767276780284
|
|
| BLAKE2b-256 |
8c5a7451c7db78ae19ef856b47c286fadb17a84269ea727e120b91d05e44d840
|
File details
Details for the file proteinmpnn-0.1.3-py3-none-any.whl.
File metadata
- Download URL: proteinmpnn-0.1.3-py3-none-any.whl
- Upload date:
- Size: 68.1 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.0.1 CPython/3.12.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ddfb45a5f80b323a1c198bbdac3d4f13dc61ddd4693eaba8059d93f6506533f8
|
|
| MD5 |
8145e7890897a2c3d516ae8c3781b9dc
|
|
| BLAKE2b-256 |
9e45b0f1661eb702f07fb694cef08d0c23d6718de23108e6844dff877da3e1e6
|







