Integer Linear Programming for Protein Library Design

Project description





Introduction
Welcome to the protlib-designer repository! This repository contains a lightweight python library for designing diverse protein libraries by seeding linear programming with deep mutational scanning data (or any other data that can be represented as a matrix of scores per single-point mutation). The software takes as input the score matrix, where each row corresponds to a mutation and each column corresponds to a different source of scores, and outputs a subset of mutations that Pareto-minimizes the scores from the different sources while maximizing the diversity of the library.
The paper Antibody Library Design by Seeding Linear Programming with Inverse Folding and Protein Language Models uses this software to design diverse antibody libraries by seeding linear programming with scores computed by Protein Language Models (PLMs) and Inverse Folding models.
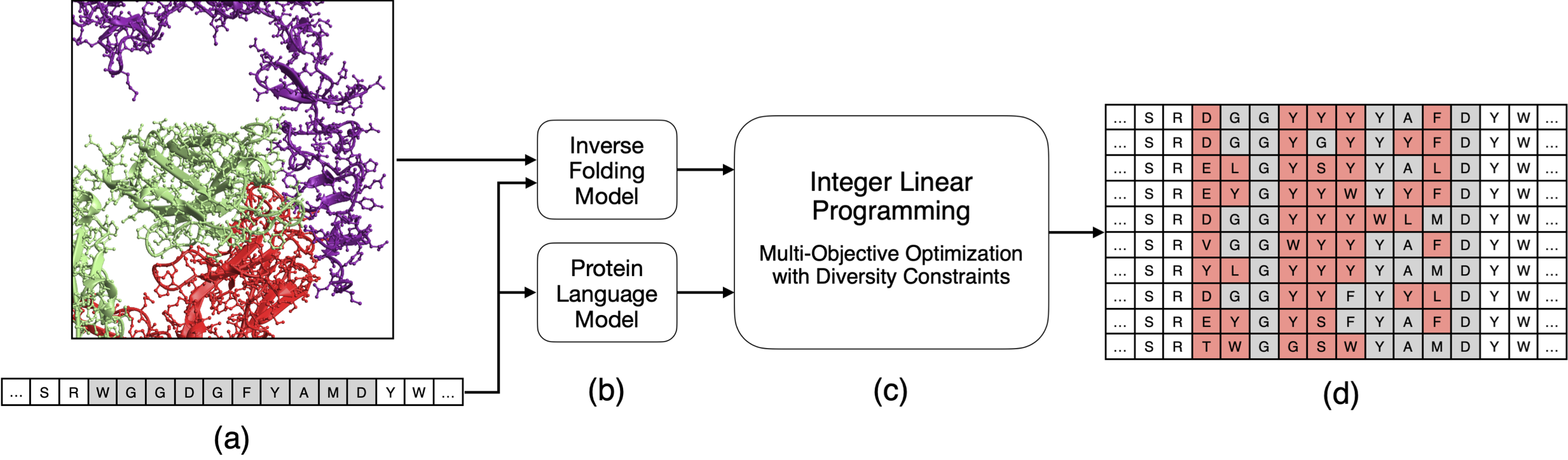
protlib-designer designs diverse protein libraries by seeding linear programming with deep mutational scanning data. (a) The input to the method is target protein sequence and, if available, a structure of the protein or protein complex (in this case, the antibody trastuzumab in complex with the HER2 receptor). (b) We generate in silico deep mutational scanning data using protein language and inverse folding models. (c) The result is fed into a multi-objective linear programming solver. (d) The solver generates a library of antibodies that are co-optimized for the in silico scores while satisfying diversity constraints.
Getting Started
In this section, we provide instructions on how to install the software and run the code.
Installation
You can pip install the package from PyPI:
pip install protlib-designer
Note: You can install all additional dependencies by running
pip install protlib-designer[all]. This will install all the dependencies for the scoring functions, including the inverse folding and protein language model dependencies.
Alternatively, you can clone the repository and install the package from source. First, clone the repository and create an environment with Python >=3.10,<3.11. Then, you can do:
python -m venv .venv
source .venv/bin/activate
pip install -e .
If you want a development environment, you can install the development dependencies:
pip install -e .[dev]
which will allow you to run the tests and the linter. You can run the linting with:
black -S -t py39 protlib_designer scripts && \
flake8 --ignore=E501,E203,W503 protlib_designer scripts
Run the code
To run the code to create a diverse protein library of size 10 from the example data, run the following command:
protlib-designer ./example_data/trastuzumab_spm.csv 10
We provide a rich set of command-line arguments to customize the behavior of protlib-designer. For example, the following command runs protlib-designer with a range of 3 to 5 mutations per sequence, enforcing the interleaving of the mutant order and balancing the mutant order, allowing for each mutation to appear at most 1 time and a position to be mutated at most 4 times,
and using a weighted multi-objective optimization:
protlib-designer ./example_data/trastuzumab_spm.csv 10 \
--min-mut 3 \
--max-mut 5 \
--interleave-mutant-order True \
--force-mutant-order-balance True \
--schedule 2 \
--schedule-param '1,4' \
--weighted-multi-objective True
For more information on the command-line arguments, run:
protlib-designer --help
Input data
The input to the software is a matrix of per-mutation scores (the csv file trastuzumab_spm.csv in the example above). Typically, the score matrix is defined by in silico deep mutational scanning data, where each row corresponds to a mutation and each column corresponds to the score computed by a deep learning model. See the example data in the example_data directory for an example of the input data format. The structure of the input data is shown below:
| Mutation | score-1 | score-2 | ... | score-N |
|---|---|---|---|---|
| AH106C | -0.1 | 0.2 | ... | 0.3 |
| AH106D | 0.2 | -0.3 | ... | -0.4 |
| ... | ... | ... | ... | ... |
| YH107A | -0.3 | 0.4 | ... | -0.5 |
| ... | ... | ... | ... | ... |
Important notes about the input data:
• The Mutation column contains the mutation in the format : WT_residue + chain + position_index + mutant_residue. For example, A+H+106+C = AH106C represents the mutation of the residue at position 106 in chain H from alanine to cysteine.
• The score-1, score-2, ..., score-N columns contain the scores computed by the deep learning models for each mutation. Typically, the scores are the negative log-likelihoods ratios of the mutant residue and the wild-type residue, computed by the deep learning model:
s_{ij}^{\text{PLM}} = -\log \left( \frac{p(x_i = a_j | w)}{p(x_i = w_i | w)} \right) = -\log(p(x_i = a_j | w)) + \log(p(x_i = w_i | w)),
where $w$ is the wild-type sequence, and $p(x_i = a_j | w)$ is the probability of the mutant residue $a_j$ at position $i$ given the wild-type sequence $w$ as estimated by a Protein Language Model (PLM) or an Inverse Folding model (or any other deep learning model). For example, in Antibody Library Design by Seeding Linear Programming with Inverse Folding and Protein Language Models, we used the scores computed by the ProtBert and AntiFold models.
Computing Input Data using Protein Language Models
We provide a set of scoring functions that can be used to compute the scores for the input data. The scoring functions are defined in the protlib_designer/scorer module. To use this functionality, you need to install additional dependencies:
pip install -e .[plm]
After installing the dependencies, you can use the scoring functions to compute the scores for the input data. For example, we can compute the scores using Rostlab/prot_bert and facebook/esm2_t6_8M_UR50D models, and then, call protlib-designer to design a diverse protein library of size 10:
protlib-plm-scorer \
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSS \
WB99 GB100 GB101 DB102 GB103 FB104 YB105 AB106 MB107 DB108 \
--models Rostlab/prot_bert \
--models facebook/esm2_t6_8M_UR50D
After that, you can run protlib-designer with the generated scores:
protlib-designer plm_scores.csv 10 --weighted-multi-objective True
Computing Input Data with Inverse Folding Models
We provide built-in scoring functions to evaluate your input structures using inverse‐folding methods. Currently, we support:
- Robust deep learning–based protein sequence design using ProteinMPNN (Dauparas et al. 2022) - Paper - We adopt some of the open source code from ProteinMPNN.
To enable inverse-folding scoring, install the extra dependencies:
pip install -e .[ifold]
Note: This will automatically download the default ProteinMPNN model weights. If you already have the weights locally, skip the download by passing
--model-pathto the scorer (see below).
Following the example in the previous section, you can compute the scores using the inverse-folding model:
protlib-ifold-scorer \
example_data/1n8z.pdb \
WB99 GB100 GB101 DB102 GB103 FB104 YB105 AB106 MB107 DB108 \
Contributing
Please read CONTRIBUTING.md for details on our code of conduct, and the process for submitting pull requests to us.
Citation
If you use this software in your research, please cite the following paper:
@article{Hayes2024.11.03.621763,
author = {Hayes, Conor F. and Magana-Zook, Steven A. and Gon{\c{c}}alves, Andre and Solak, Ahmet Can and Faissol, Daniel and Landajuela, Mikel},
title = {Antibody Library Design by Seeding Linear Programming with Inverse Folding and Protein Language Models},
journal = {bioRxiv},
year = {2024},
elocation-id = {2024.11.03.621763},
doi = {10.1101/2024.11.03.621763},
publisher = {Cold Spring Harbor Laboratory},
url = {https://www.biorxiv.org/content/early/2024/11/03/2024.11.03.621763},
eprint = {https://www.biorxiv.org/content/early/2024/11/03/2024.11.03.621763.full.pdf}
}
License
protlib-designer is released under an MIT license. For more details, please see the
LICENSE and RELEASE files. All new contributions must be made under the MIT license.
SPDX-License-Identifier: MIT
LLNL-CODE-2001645
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file protlib_designer-2.0.1.dev13.tar.gz.
File metadata
- Download URL: protlib_designer-2.0.1.dev13.tar.gz
- Upload date:
- Size: 1.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.10.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
332c68e4decc92acf0ca48e3bc5ba0ad83f57fd8133587ed3a942d617657510e
|
|
| MD5 |
ff571674c14f6f5fd88403cf4b617f33
|
|
| BLAKE2b-256 |
25a0e5b506103c9a43f03019b2dc3a237f1bd6e8a62aa1c3cb21ca05d341acd9
|
File details
Details for the file protlib_designer-2.0.1.dev13-py3-none-any.whl.
File metadata
- Download URL: protlib_designer-2.0.1.dev13-py3-none-any.whl
- Upload date:
- Size: 17.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.10.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
68861069f2a79f89f0c5a7a8b1d27f21a7688959adbab87ce65a808f3205be4b
|
|
| MD5 |
0052d31d3468fe7ecd60702fdf000be9
|
|
| BLAKE2b-256 |
a30951c2b4f1a2a259e734bde6aa9349345d2d971a6f90dc360405270378906d
|







