selectivity prediction of electrophilic substitutions on (hetero)aromatic compounds

Project description
RegioSQM
Background
RegioSQM predicts the (hetero)aromatic CH sites most likely susceptible to the electrophilic aromatic substitution (EAS). For this, the heat of formation of protonated intermediates is computed by MOPAC at the PM3/COSMO level.1 When testing this approach for 535 substrates belonging to 69 groups (e.g., benzenes, pyridines, pyridones), the authors observed 96% of the computed predictions to match the experimental evidence. The authors maintain a dedicated web site, regiosqm.org, to perform these computations for individual molecules, expressed by a SMILES string.2
With the scripts of this repository, RegioSQM may be used locally. RegioSQM then may be used for the serial prediction about substrates expressed as a list of SMILES strings. Most of the information provided here is based on the seminal RegioSQM paper, an open access publication.
Show case
For a given substrate, RegioSQM probes any (hetero)aromatic position theoretically susceptible for an electrophilic substitution reaction (EAS) by the addition of hydrogen to yield a charged intermediate. For each position, the heat of formation of this intermediate is computed. As for pyrazole (1, line a), for example,
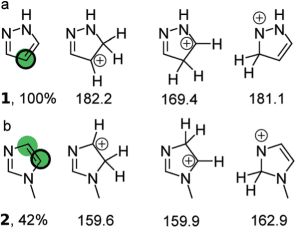
protonation in the 4-position yields the least endothermic charged regioisomer (169.4 kcal/mol, if computed at the level of PM3/COSMO)3 which RegioSQM indicates by a green dot. This is backed by experimental findings; in the course of an EAS, bromine of N-bromosuccinimide (NBS) exclusively adds to this position. The illustrations indicate the sites experimentally determined as most prominent to the EAS by a black circle.
The site RegioSQM predicts as most susceptible to the EAS serves as a reference. RegioSQM then compares the heat of formation about regioisomers of this reference intermediate. If the heat of formation about the test site's intermediate differs by less than 1 kcal/mol (4.18 kJ/mol) from the one about the reference site, RegioSQM marks the test site equally by a green dot. The site is marked red if the difference with the reference site is more 1 kcal/mol, but less than 3 kcal/mol (12.6 kJ/mol). RegioSQM's prediction about N-methyl imidazole (2, line b) is backed by experimental evidence; indeed, the EAS with NBS yields a mixture by preferential reaction at the two positions highlighted.
If RegioSQM recognizes a substrate as conformational flexible, by default, per site theoretically susceptible to an EAS, the heat of formation about up to 20 conformers of the intermediate are computed. The prediction then ranks the least endothermic charged conformer per site.
As shown, sometimes, the experimentally observed sites of EAS (black circle) are not those RegioSQM predicts as highly susceptible (green dot) or moderately susceptible (red dot) to the EAS. Steric hindrance to entrant electrophiles, for example, is not considered by RegioSQM's prediction yet may be one plausible cause for such a discrepancy. This however should be balanced with the low computational cost of the method deployed (PM3/COSMO instead of DFT) to predict rapidly the sites of the EAS reaction. Depending on the threshold used, the rate of success within the test set of 535 substrates equals to 92% or 96%.
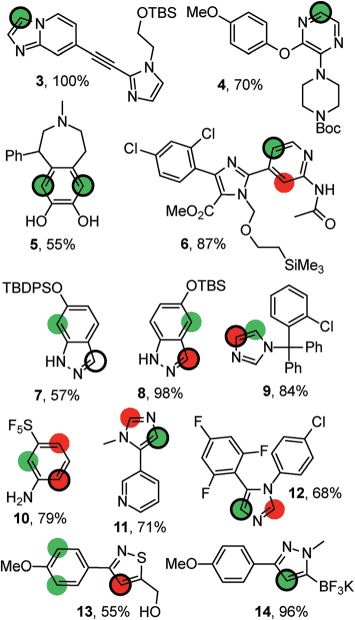
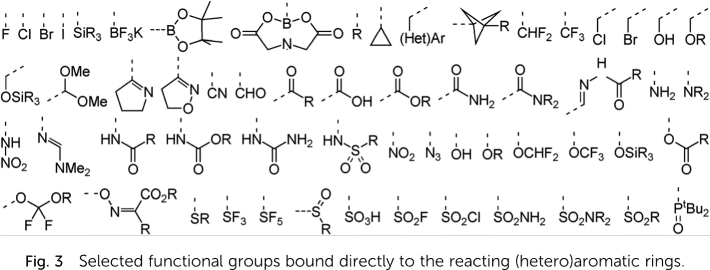
Data in subfolder replication permit a replication of this prediction
for 535 substrates obtained by permutation of 69 mono- and bicyclic
(hetero)aromatic core structures with the substituents like those
depicted below:
Proposed deployment
Local installation
The overall analysis depends on the freely available opensource MOPAC to perform quantum chemical computations. Its download page provides installers for Linux, Mac, and Windows. You may consult repology.org to check if your distribution provides a package (example Linux Debian), too.
RegioSQM is a collection of Python scripts and modules organized around
a pyproject.toml file. Its dependencies
(NumPy,
RDKit,
openbabel) are most comfortably
resolved within a virtual environment. Thus, check the release page of
this repository for a .whl of regioSQM (about 20kB). If not present,
or if the commit history of the main branch of this repository advanced
vs the publication of a .whl, you equally can create a .whl on your
own, for instance by either one of
python -m build # see https://pypi.org/project/build/
uv build # see https://docs.astral.sh/uv/concepts/projects/build/
with a copy of this GitHub archive. A third option is to cd into the decompressed archive (about 23MB) and run a
pip install .
In an instance of Linux Debian 14/forky (branch testing with Python 3.13.14), the size of the supporting virtual environment is about 285MB.
Local use
The successful installation of regioSQM provides the regiosqm command
to your CLI. This includes a minimal help menu with prompts
(regiosqm -h).
-
preparation of the input file
Your input file is a list of an alphanumeric identifier of your structure of interest followed by a SMILES string. The two columns are whitespace separated. An example snippet is
benzene c1ccccc1 pyridine c1ccncc1 comp402 c1c(n(cc1)C1COC1)C=O comp437 c1ccc(o1)Sc1ccccc1 comp413 c1ccc(s1)/C=N/N=C/c1sccc1 comp1 n1ccc[nH]1
A suggestion is to name this file
input.smi; however, regioSQM does not constrain you on your choice of file name, nor file extension. Programs like Avogadro2 and OpenBabel may help you to obtain, or – by conversion from other files – assign a SMILES string to populate your list. -
generation of conformers
Presuming an input file by name of
input.smi, the commandregiosqm -g input.smi > conformes.csv
writes for up to 20 confomers per input SMILES string a MOPAC input file (
.mop). If wanted, this upper threshold can be adjusted (see flag-c/--max_conformations).A typical MOPAC input file generated (here an example about benzene) looks like
pm3 charge=1 eps=4.8 cycles=200 C 1.47640 1 0.40320 1 0.32980 1 C 0.58630 1 1.35550 1 0.11090 1 C -0.79350 1 1.02470 1 -0.19620 1 C -1.33460 1 -0.34490 1 -0.29440 1 C -0.23540 1 -1.29390 1 -0.02810 1 C 1.00490 1 -0.97140 1 0.24740 1 H 2.47770 1 0.69830 1 0.55160 1 H 0.88480 1 2.39810 1 0.16000 1 H -1.47920 1 1.86660 1 -0.36680 1 H -1.72440 1 -0.45340 1 -1.33180 1 H -2.12910 1 -0.54760 1 0.45240 1 H -0.47390 1 -2.36820 1 -0.06310 1 H 1.74010 1 -1.76710 1 0.42840 1
The setup with a dielectric constant of 4.8 corresponds to chloroform at a temperature of 20 Celsius4 (293 K). On your own risk you can change the dielectric constant for instance to 37.3 (as for nitromethane, a value equally compiled by Alfa Chemistry) for instance by
sed -i 's/eps=4\.8/eps=37.3/g' *.mop
File
conformers.csvtracks the sites to be tested for the electrophilic substitution for every conformer of every input SMILES string provided, e.g.name, SMILES, reaction_center, len(conformations) benzene+_1, C1=C[CH+]CC=C1, 0, 1 , charge=1 benzene+_2, C1=C[CH+]CC=C1, 5, 1 , charge=1 benzene+_3, C1=C[CH+]CC=C1, 1, 1 , charge=1 benzene+_4, C1=C[CH+]CC=C1, 2, 1 , charge=1 benzene+_5, C1=C[CH+]CC=C1, 3, 1 , charge=1 benzene+_6, C1=C[CH+]CC=C1, 4, 1 , charge=1
-
work with MOPAC
In the overall analysis, especially with larger / more flexible molecules and datasets, the computation with MOPAC is the rate limiting step. Thus, it is recommended to parallelize the work with the
.mopfiles. With GNU Parallel (entry on repology.org, example package of Linux Debian), a command likels *.mop | parallel -j4 "mopac {}"
runs up to 4 concurrent processes of MOPAC. Depending on the number of CPU cores at your disposition, you may adjust flag
-jto your preference.A less efficient sequential run supported by Debian's BASH could be
for file in *.mop do mopac "$file" done
If you work with Windows with access to Windows git and its Git Bash, the functionally equivalent command would be
for file in *.mop; do mopac.exe "$file"; done
-
analysis of the results
Back in regioSQM, the call of
regiosqm -a input.smi conformers.csv > results.csv
analyzes MOPAC's results. Per submitted SMILES string, the tally reports the predictions about an aromatic electrophilic substitution. In the .svg simultaneously generated, green disks highlight the most favorable site(s), red disks somewhat favorable site(s).
Note that the result of the prediction may depend on the usage of MOPAC. A more obvious reason is the aforementioned change of the dielectric constant, a less obvious one the release of MOPAC used.
Extensive check
Further development of MOPAC and RegioSQM may affect the prediction of
sites deemed exceptionally susceptible to the EAS reaction. To identify
changes since submission of the seminal publication in 2017, the
scrutiny of substrates tested was replicated with MOPAC 2016
(version 20.173L, 64-bit). Tools used and intermediate results obtained
(e.g., SMILES strings / illustrated atom indices per EAS class) as
obtained with release 2.0.0-beta are provided in folder replication.
Especially the results in sub-folder predicted_sites allow a quick
comparison of a current and of future local installations of RegioSQM a
rapid diffview of texts.
In comparison of the results depicted in the SI of the seminal paper, only 47 out of 535 pattern (8.8%) reexamined changed since them. Among these, changes for the definitively better (22 pattern, about 4.1%) or definitively worse (22) are scattered over multiple EAS classes. For 2 pattern (about 0.4%), no attribution for the better or worse was made.
Footnotes
-
MOPAC's use of COSMO, the «COnductor-like Screening MOdel» by Klamt and Schüümann is described in MOPAC's manual. By default, computations by RegioSQM are performed with MOPAC's implicit effective van der Waals radius of the solvent of 1.3 and an explicitly defined dielectric constant of 4.8 (chloroform, see module
molecule_formats.py, line 62). ↩ -
If your molecule sketcher of choice does not offer the export into this format, consider OpenBabel for a (batch) conversion of your structure files into this format, or copy-paste the strings provided by a service like the PubChem Sketcher. ↩
-
MOPAC's use of COSMO, the «COnductor-like Screening MOdel» by Klamt and Schüümann is described in MOPAC's manual. By default, computations by RegioSQM are performed with MOPAC's implicit effective van der Waals radius of the solvent of 1.3 and an explicitly defined dielectric constant of 4.8 (chloroform, see module
molecule_formats.py, line 62). ↩ -
For a compilation of dielectric constants, see for instance the compilation on https://www.alfa-chemistry.com/resources/table-of-dielectric-constants-of-liquids.html ↩
Release history Release notifications | RSS feed

Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file regiosqm-3.0.0.tar.gz.
File metadata
- Download URL: regiosqm-3.0.0.tar.gz
- Upload date:
- Size: 17.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b187af8bc009e2eae193171c4d062092b9a0fea470c1b6c450a6f320649b71e3
|
|
| MD5 |
234fc3e32a17cc555160c6905a0a820b
|
|
| BLAKE2b-256 |
55015990e68050b9ff98238dd650478a6e908464835dfbb5732d2ac568fd573f
|
Provenance
The following attestation bundles were made for regiosqm-3.0.0.tar.gz:
Publisher:
publish-to-pypi.yml on nbehrnd/RegioSQM
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
regiosqm-3.0.0.tar.gz -
Subject digest:
b187af8bc009e2eae193171c4d062092b9a0fea470c1b6c450a6f320649b71e3 - Sigstore transparency entry: 2102197685
- Sigstore integration time:
-
Permalink:
nbehrnd/RegioSQM@a14edf0288aa37113fdf605b48f114bf400c5b28 -
Branch / Tag:
refs/heads/main - Owner: https://github.com/nbehrnd
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish-to-pypi.yml@a14edf0288aa37113fdf605b48f114bf400c5b28 -
Trigger Event:
workflow_dispatch
-
Statement type:
File details
Details for the file regiosqm-3.0.0-py3-none-any.whl.
File metadata
- Download URL: regiosqm-3.0.0-py3-none-any.whl
- Upload date:
- Size: 18.3 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5a7992ef9dd85c61db5005eb9f00bdf18352764c97a63d16d8f4edd337589259
|
|
| MD5 |
7125bcffa44223c7c5f4991380773c8f
|
|
| BLAKE2b-256 |
1812de1c4e46041d7aadf77e62f8af892e57dbd9ce344521dcf5e294e07657ce
|
Provenance
The following attestation bundles were made for regiosqm-3.0.0-py3-none-any.whl:
Publisher:
publish-to-pypi.yml on nbehrnd/RegioSQM
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
regiosqm-3.0.0-py3-none-any.whl -
Subject digest:
5a7992ef9dd85c61db5005eb9f00bdf18352764c97a63d16d8f4edd337589259 - Sigstore transparency entry: 2102197872
- Sigstore integration time:
-
Permalink:
nbehrnd/RegioSQM@a14edf0288aa37113fdf605b48f114bf400c5b28 -
Branch / Tag:
refs/heads/main - Owner: https://github.com/nbehrnd
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish-to-pypi.yml@a14edf0288aa37113fdf605b48f114bf400c5b28 -
Trigger Event:
workflow_dispatch
-
Statement type:







