A toolbox for protein engineering and DNA shuffling

Project description
Software
Welcome to seq-mutator protein engineering tool by the iGEM Team of the University of Münster 2024.
[[TOC]]
Abstract
Protein engineering is a critical field that allows for the design and modification of proteins with enhanced or novel functions, driving innovations in medicine, biotechnology, and environmental sustainability. Testing protein variants is labor expensive so using computer aided methods to predict promising candidates has been of high interest for decades. Novel AI technology opens up promising new capabilities that can be leveraged to enhance previous techniques for sequence-function predictions. However, they are often data-intensive and require specialist knowledge for choosing and using appropriate models.
Since there is no high throughput assay available to test the activity of our enzyme, we were very limited in the number of variants we were able to test. That is why we chose a modeling method which can predict highly functional variants with only a low number (>=24) of measurements 1.
Additionally, to complete our protein engineering toolbox, the software includes a DNA sequence similarization and shuffling tool. Such tools already existed in the past but were not accessible anymore. Our tool performs the usual codon optimization and similarization for a set of proteins, but also includes restriction sites from a list of available restriction enzymes, thus also making AI-based low N restriction enzyme mediated DNA shuffling affordable for iGEM Teams, other researchers, or smaller companies.
Since we suspect many other iGEM teams will be facing the same challenge of limited measuring capability, we implemented a user friendly cli software tool, allowing them to easily have access to data efficient protein engineering tools. Our software tool supports current state-of-the-art protein language models, multi-GPU training, memory optimization techniques like LoRa (Low Rank Adaption), and more, making our tool a highly accessible one-stop solution for AI-driven protein engineering.
Description
Low-N Protein Engineering
To find highly active protein variants we adapted an approach introduced by Biswas et. al. in 1. This approach leverages a protein language model extracting so called features of the input sequence and then uses measured activities mapping those features to actual activities.
We introduced a novel implementation for this method using a more modern basemodel and adapted more advanced methods for training transformer models.
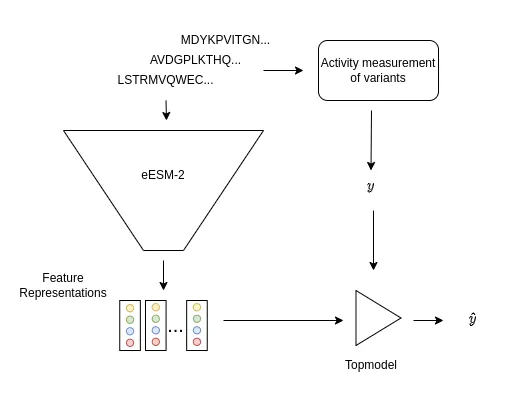
Figure 1: A set of variants of the target protein is selected and the enzymatic activities are measured (y). The fine-tuned ESM-2 model (eESM-2) receives those variant sequences as input and generates feature representations of those variants. Those representations and the activities are used to create a topmodel linking the feature representations to protein activities.
The method makes use of a fine-tuned (evotuned) protein language model to generate feature representations of those proteins. The method then requires the selection of a variant library of the target protein. This library can be selected by a combination of rational design, zero-shot prediction or random selection.
Note: Based on our investigation on the performance of different training set designs we recommend testing protein variants chosen based on the zero-shot prediction (modeling page).
The measured activities of those variants and the feature representations are then used to train a linear regression topmodel (Fig 1.). The regression topmodel then predicts activities of unseen variants. To generate a promising variant with a combination of mutation the method uses a in silico directed evolution algorithm 1 to allow high-throughput screening even when experimental assays are expensive or not available.
To read more about the background and how we applied this method for our modeling approach please visit our modeling site.
Unlike many similar tools that rely on loosely coupled Jupyter notebooks, often sacrificing readability and adaptability, our software tool, seq-mutator, provides a seamless, all-in-one solution. It integrates distinct modules for each step of the low N protein engineering workflow, enabling AI-guided protein design with greater efficiency and clarity:
search: Creating a library of evolutionary related sequences for a given target protein.evotune: Fine-tuning a protein language model with given amino acid sequences.topmodel: Training a topmodel on given variant activities and predicting activities for given sequences.directed evolution: Building new protein variants with several mutations for a protein target given a trained topmodel.
Zero-Shot Prediction
The same (optionally evotuned) ESM-2 model can be used for fitness prediction without any experimental data at all, so called zero-shot inference 2. We incorporated this method into our tool, since it can be useful to get an initial overview of beneficial mutations. It can also be beneficial to use zero-shot prediction results when creating the training set for the low N topmodel, as we saw in our in-silico experiments, as training sets designed from zero-shot predictions appear to be more informative than random library generation by error-prone PCR. Those results, a more detailed explanation of the zero-shot method, and recommendations on informative training set design can be found on our modeling site.
DNA Shuffling
Another method we wanted to adopt for our protein engineering approach is DNA shuffling. Our tool allows us to enter two Protein sequences and output DNA sequences with an improved sequence overlap. This improves the shuffling efficiency of Proteins with comparably low sequence identity. Such tools have been published in the past, but none of the more elaborate tools are accessible anymore 3. Further, we extended our tool to support Restriction Enzyme-Mediated DNA Shuffling, a technique described by Gillam et al 4. This approach allows the restriction and combination of certain fragments, thus allowing for cheap generation of designed enzyme chimeras (Fig. 1). Such an information-based approach can reduce the number of variants that must be tested and driven by our low N model. Our shuffling software integrates unique restriction sites in the overlapping regions of the coding sequences of the proteins.
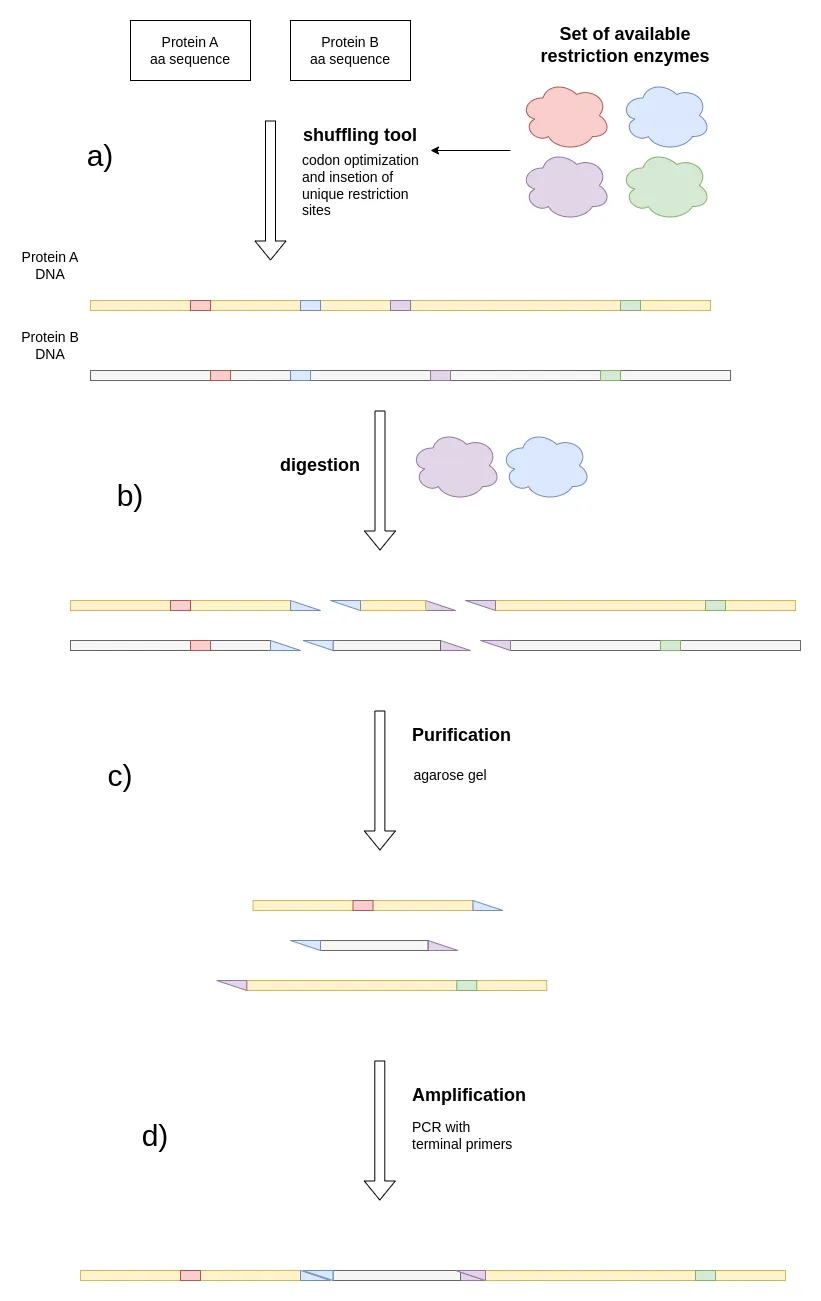
Figure 2: Shows the process of Restriction Enzyme-Mediated DNA Shuffling. a), two proteins with at least ~65% amino acid overlap 4 that are to be shuffled are obtained and a list of available restriction enzymes is obtained. The shuffling tool performs codon optimization while placing unique restriction sites of the given enzymes into the DNA sequence. b), a digestion with 2 restriction enzymes is performed, cutting the sequences into fragments. c), certain fragments are purified (e.g. via agarose gel). d) the fragments are ligated to create the desired protein chimeras.
One challenge with Restriction Enzyme-Mediated DNA Shuffling is the expense associated with purchasing restriction enzymes. Large companies often have an extensive library of these enzymes, but iGEM teams may face difficulties affording new ones. Our shuffling software addresses this issue by utilizing a list of enzymes that are already available in your lab’s inventory. The tool selectively incorporates only those restriction sites. Overall, our tool can help to reduce the cost and time for creating desired chimeras by massively reducing the need for DNA synthesis.
Designing training sets for low N protein engineering with DNA shuffling, explores large areas of sequence space, potentially allowing a higher trust radius for mutations introduced during the in silico directed evolution phase.
Use Case: Engineering the Piperamide Synthase
In our goal to develop a feasible bioproduction of a mosquito repellent called NBP (N-benzoylpiperidine). We tried to combine several enzymes to build an enzymatic cascade to produce NBP out of the amino acids Lysine and Phenylalanine (Fig. 3).
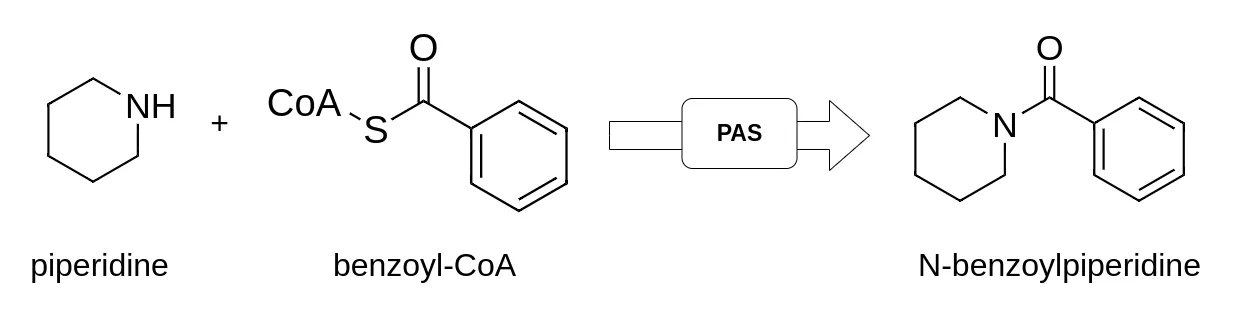
Figure. 3: The reaction of Benzoyl-CoA and Piperidine to the mosquito repellent N-benzoylpiperidine (NBP) catalyzed by the piperamide synthase (PAS).
However, the last enzyme in our designed metabolic pathway, the piperamide synthase (PAS) from Piper Nigrum that catalyzes the reaction from Benzoyl-Coa and Piperidine to NBP, had a low catalytic activity. To increase the performance of the PAS in this limiting step in the cascade, we engineered the PAS and analyzed its catalytic activity via HPLC-MS. We made heavy use of seq-mutator for finding new promising PAS variants.
Therefore, we used the zero-shot method implemented in this tool, LigandMPNN and rational design to successfully generate a library of 28 PAS variants. After cloning using QuickChange PCR, expression in E. coli and enzyme purification, we performed an in vitro activity assay to test the NBP yield of each variant.
For our basemodel, we fine-tuned the ESM-2 model with 650M parameters with the evotuning tool of seq-mutator. For that we used ~70 000 sequences obtained by the search tool of seq-mutator.
We used the measured activities and the fine-tuned basemodel to train the topmodel and the in silico directed evolution algorithm implemented in seq-mutator to generate new promising variants of the PAS with 1, 3 or 7 new mutations. The results of those experiments can be found on our modeling site.
Implementation
We used python3 to implement a user-friendly cli combining the shuffling tool and the low N protein engineering to create a one-stop protein engineering toolbox called seq-mutator for accessible AI-driven protein engineering.
The shuffling makes use of the widely used Python library BioPython. The restriction enzymes are parsed using BioPython, which fetches a registry of restriction enzymes from external sources. Therefore, if new restriction enzymes emerge, they will also be available in our software tool. To input the proteins the FASTA format is used and for the codon table and the restriction enzymes the CSV format is used.
The low N protein engineering tool is developed in python3 and makes heavy use of the Hugging Face library. By using this standard platform for working with transformer models like ESM-2 we enable effortless integration with multi-GPU training, LoRa performance optimization and standardized logging with tensorboard. Therefore, future novel protein language models will automatically be available for our software tool and the tool will be easily extensible for new methods that are developed for transformer models. Users only need to provide a FASTA file of their engineering target and a CSV file containing their measured sequence-activity data.
Installation
Prerequisites
The software tool is only tested for the following Python versions:
- Python 3.10.11
- Python 3.10.12
Python3 Installation
Here is how you install Python on different operating systems:
For windows you might need to add the python installation to your PATH to make the python3 command available system wide. You can check out the following guide to learn how to do this.
On Linux you can add an executable to your PATH by running the following command in your terminal:
echo "export PATH=$PATH:/path/to/your/executable" >> ~/.bashrc
. ~/.bashrc
You can do the same on MacOS by running:
echo "export PATH=$PATH:/path/to/your/executable" >> ~/.bash_profile
. ~/.bash_profile
Virtual Environment
We recommend using a Python3 virtual environment to install the tool. A Python3 virtual environment is an isolated environment that allows you to install and manage dependencies separately from the global Python installation, ensuring project-specific packages and versions without affecting other projects or system-wide settings.
Here is how you create a virtual environment on Ubuntu Linux / MacOS:
Note: You might need to install the
python3-venvpackage first. You can do this by running the following command in your terminal:sudo apt-get install python3-venv
Then you can create and activate a virtual environment by running:
python3 -m venv venv
source venv/bin/activate
After activating the virtual environment, you can install the tool as described below. Now all dependencies will be installed in the virtual environment and will not affect the global Python installation. After you are done working with the tool, you can deactivate the virtual environment by running:
deactivate
Here are some guides on how to setup your python environment on other operating systems:
If you do not want to use a virtual environment, you can install the tool globally. However, this is not recommended as it might affect other projects or system-wide settings.
Pip
You can install the package via pip:
pip install seq-mutator
Note: This may take a while.
If done correctly, the seq_mutator executable should be available in your PATH.
seq_mutator --help
The above command should show you the help message of the tool.
Optionally on Linux you can install autocompletion capabilities for the tool:
seq_mutator --install-completion
After restarting your terminal, you should be able to use autocompletion for the tool.
If you want to use the search tool
For performing the evotuning, a CSV file with a list of evolutionary related amino acid sequences is required. To generate this file, you can use the build in search tool, which makes use of the jackhmmer tool 5. Please install jackhmmer under the following link before using the search tool.
Note: The
jackhmmertool is only available for Linux systems. If you are on Windows, you can use the Windows Subsystem for Linux to installjackhmmer.
Make sure that then jackhmmer is in your PATH. Check out the guide above on how you can achieve that. You can check this by running the following command in your terminal:
jackhmmer -h
To perform the search, you need to have a protein database available in the FASTA format. You can download a Uniprot database for this purpose.
Alternatively, to our search module you can also use online tools like Blast for finding evolutionary related sequences to your target sequence. To use them for evotuning you must upload a CSV file containing the rows id (ID of the protein), sequence (sequence of the protein) and evalue (the E-value regarding the target protein) to the projects folder.
Note: Parsing Blast output files was not implemented due to time constraints but might be implemented in future releases.
GPU training
The tool will automatically detect CUDA viable GPUs and use them.
VRAM requirements
Using the ESM-2 model with 650 million parameters with LoRA memory optimization 6 and a maximum sequence length of 1024 we could train the model on an A100 SXM with 80 GB VRAM with a batch size of 2.
You can estimate your hardware requirements for the protein language model you want to use with Hugging Face tool.
Usage
Before you start, please create a working directory where you want to store input files, temporary files and output files. You can do this by running the following command on Linux, Windows PowerShell or MacOS terminal:
mkdir my_workspace
cd my_workspace/
mkdir data/
To use the tool, open the terminal and type the tool name seq_mutator followed by the subcommand you want to use (e.g., seq_mutator command --option value). You can always get help by using the --help flag:
seq_mutator --help
This will list further subcommands and options you can use. Each subcommand and option will be accompanied by an explanation and the default values used.
Low N Protein Engineering
The data for the Low N Protein Engineering tool is organized in databases and projects. The databases contain the protein sequences used for the search. The projects contain the input and output files for the evotuning and topmodel training. The tool will automatically create the necessary directories and files for you. The data is organized as follows:
.
└── data # data directory
├── databases # databases directory
│ └── sprot # database name
│ ├── db.fasta # original database fasta file
│ └── db.sqlite # database after building
├── models--facebook--esm2_t6_8M_UR50D/ # cached hugging face model
└── projects # projects directory
└── test # project name
├── hmmer.out # result of jackhmmer search
├── runs # runs directory containing evotuning runs
│ ├── run1/ # run name
│ └── test_run # run name
│ ├── checkpoint-3/ # model checkpoint at step 3
│ ├── checkpoint-11/ # model checkpoint at step 11
│ └── logs/ # tensorboard logs
├── sequences.csv # csv file with evolutionary related sequences
├── activities.csv # csv file with measured activities
├── topmodel.pkl # pkl file with topmodel state
└── target.fasta # target protein fasta file
Note: You can find example data in the
datadirectory of this repository. You can use this to test the software on your device. It includes an example project for thelow-nmethod and a codon table for e. Coli, a CSV file containing restriction enzymes and a protein target for theshufflemodule. You can download it from the following link. In there you can find an example project calleddefaultin thedatadirectory. It contains thetarget.fastafor the PAS as well as a search result (sequences.csv) and some example measurements (activities.csv) which you can copy into the project you will create if you need them.
Creating a project
Before you start you need to create a new project for your protein target. The following command will create a project with the name my_enzyme and will attach the given FASTA file containing the target amino acid sequence to it.
seq_mutator low_n projects create my_enzyme --target /path/to/your/enzyme_target.fasta
to see all available options run:
seq_mutator low_n projects create --help
You can validate the creation of the project by running:
seq_mutator low_n projects list # shows a list of projects
to see all available options on how to manage your projects (create, list, delete, etc.) run:
seq_mutator low_n projects --help
Advanced: Per default the tool will use the environment variable
LOWN_CURRENT_PROJECTto determine the current project and the environment variableLOWN_PROJECTS_PATHto determine the path for thedatadirectory. You can set these variables in your.bashrcor.bash_profilefile to make the tool more convenient to use. You can also create a.envfile in your working directory and set the variables there in the formatVAR_NAME=VALUE. The tool will automatically load the variables from the.envfile.
Creating and building a database
With the implemented search you can find evolutionarily related sequences for your target protein. The search is done on a protein database, so install one first. To make the databases reusable in your workspace across projects you can create a database:
seq_mutator low_n databases add db_name /path/to/your/protein_database.fasta
You can validate the database creation by running:
seq_mutator low_n databases list # should show your database name
To make the database easily searchable, for the search it is required to build the database:
seq_mutator low_n databases build db_name
Searching for evolutionarily related sequences
Before you continue make sure you have created a project and that you have a database available and built. You can search for evolutionarily related sequences by running:
This will perform a
jackhmmersearch on the database and store the results in the project directory. The search makes 5 iterations, uses 4 threads and only selects sequences with an E value <= 0.5.
seq_mutator low_n search db_name my_enzyme --num-iters 5 \
--max-evalue 0.5 \
--num-threads 4
Evotuning (fine-tuning) a Protein Language Model
Before you continue make sure that the sequences.csv is included in your project (either run the search as described above or get it from some other source). If you want to use GPU for training, make sure you have a CUDA viable GPU available. This can be checked in your terminal with the nvidia-smi command. The GPU will be automatically detected. The tool will try to use all the available GPUs.
seq_mutator low_n evotune --epochs 60 \ # training for 60 epochs
--project-name my_enzyme \ # project name
--batch-size 32 \ # batch size
--take 0.1 \ # take 10% of the sequences
--eval epoch \ # evaluate on the validation set after each epoch
--model "facebook/esm2_t6_8M_UR50D" \ # use the ESM-2 model with 8M parameters
--tokenizer "facebook/esm2_t6_8M_UR50D" \ # use the ESM-2 tokenizer
--run "test_run" \ # run name under which logs and checkpoints will be stored
--max-length 1024 \ # maximum sequence length
--lora # use the LORA model
Per default variable batch size is activated. So, if the batch size you picked is too high for your GPU memory, the tool will automatically reduce the batch size.
During training, the tool will automatically save model checkpoints and logs into the project directory. You can monitor the training process by launching tensorboard:
tensorboard --logdir my_workspace/data/projects/my_enzyme/runs/test_run/logs
You can also continue training from a checkpoint by running:
seq_mutator low_n evotune --epochs 60 \ # training for 60 epochs
--project-name my_enzyme \ # project name
--batch-size 32 \ # batch size
--take 0.1 \ # take 10% of the sequences
--eval epoch \ # evaluate on the validation set after each epoch
--model "/path/to/checkpoint" \ # use the desired checkpoint
--tokenizer "facebook/esm2_t6_8M_UR50D" \ # use the ESM-2 tokenizer
--run "test_run" \ # run name under which logs and checkpoints will be stored
--max-length 1024 \ # maximum sequence length
--lora # use the LORA model
The number behind the checkpoint folders is the step number. You can find the step number in the tensorboard logs.
For further analysis of the log data, we recommend the tool tbparse for parsing the tensorboard logs into a pandas dataframe.
Topmodel
Before you train your topmodel make sure that the activities.csv with your measurements are included in your project. You can train a topmodel by running:
seq_mutator low_n topmodel train --project-name my_enzyme \ # project name
--model "facebook/esm2_t6_8M_UR50D" \ # use the ESM-2 model with 8M parameters
--tokenizer "facebook/esm2_t6_8M_UR50D" # use the ESM-2 tokenizer
If you want to use a fine-tuned model, you can specify the checkpoint path:
seq_mutator low_n topmodel train --project-name my_enzyme \ # project name
--model "/path/to/your/checkpoint" \ # use the desired checkpoint
--tokenizer "facebook/esm2_t6_8M_UR50D" # use the ESM-2 tokenizer
You can then use the topmodel to predict the activity of an input sequence:
seq_mutator low_n topmodel predict "MAAAK" --project-name my_enzyme
The first argument is the sequence you want to predict the activity for. It can be a single sequence or a path to a FASTA or CSV file containing amino acid sequences.
Zero-Shot prediction
The zero-shot prediction can be used to score single point mutations without any experimental or mechanistic knowledge on your target protein, either all ones or a subset thereof. With the tool, you can create a CSV file containing all mutations:
seq_mutator low_n zero_shot init --project-name my_enzyme
Note: The tool will automatically create a CSV file containing all possible point mutations for your target protein in the
projects/<project_name>folder. Heredmsstands for deep mutational scann.
The zero-shot prediction scores all point mutations inside the point_mutation_data.csv.
seq_mutator low_n zero_shot predict --model "facebook/esm2_t6_8M_UR50D" --project-name my_enzyme --mutation-col mutant
Be aware that zero-shot predictions are based on likelihoods of amino acids as predicted by protein language models. In general, these likelihoods are derived from evolutionary data that the protein language models learned during their training 2. Zero-shot prediction scores migth therefore not correlate with your desired activity, which often has a different objective than what evolution might act upon. It is, therefore, advised to carefully analyze targets identified by zero-shot predictions. Nevertheless, we analyzed that zero-shot predictions can have a high informational value for the performance of the topmodel and we thus recommend characterizing variants designed by zero-shot predictions instead of random sampling. You can further read about the zero-shot method and its application in our modeling site.
Directed evolution
During in silico directed evolution to the low N protein engineering workflow, protein variants are genereated and their activity is predicted by the topmodel, which has been trained on the data of >= 24 variants of your target protein. Therefore, the topmodel.pkl must exist in the project folder before starting the in silico directed evolution (see above). Then we can run the evolution algorithm:
seq_mutator low_n directed_evolution run --model "facebook/esm2_t6_8M_UR50D" --project-name my_enzyme --n-iterations 100
Restriction Enzyme-Mediated DNA Shuffling
In case users want to design variants with DNA shuffling techniques, the codon optimization with restriction site insertion is performed in two steps. In the first step, restriction sites in the overlapping regions are identified and stored as CSV a file. The user can then manually check the restriction sites.
In the second step, the codon optimization is performed using the updated CSV file with the restriction sites.
The tool outputs the optimized DNA sequences as stdout. You can redirect the output to a file by using the > operator.
Finding restriction sites
This command requires 3 arguments:
--alignment: the path to the alignment file in FASTA format containing proteins with at least ~65% sequence identity--codon-table: the path to the codon table file in CSV format. This codon table is unique for the organism you are working with. Codon tables for commonly used organisms are provided in this repository atdata/shuffling/.--restriction-enzymes: the path to the restriction enzymes file in CSV format. This file contains the restriction enzymes the tool should try to include into the sequence.
You can run the command as follows:
seq_mutator shuffle scan --alignment /path/to/alignment.fasta \
--codon-table /path/to/codon_table.csv \
--restriction-enzymes /path/to/restriction_enzymes.csv
This command will output a hits.csv CSV file containing the restriction sites found in the alignment.
Please review them and save the file.
Codon optimization
In the next step the codon optimization can be performed given the hits.csv file:
seq_mutator shuffle optimize --alignment /path/to/alignment.fasta \
--codon-table /path/to/codon_table.csv \
--restriction-enzymes /path/to/restriction_enzymes.csv \
--hits /path/to/hits.csv
The tool will output the optimized DNA sequences as stdout. You can redirect the output to a file by using the > operator.
Before ordering the sequence, you should further validate the sequences by checking the restriction sites. You should also use following IDT tools to validate if the sequence is fit to be synthesized.
Contributing
If you want to contribute to the project, please follow the following steps:
- Fork the project
- Open an issue and describe the feature you want to implement or the bug you want to fix
- Create a new branch with the issue number and a brief description of the issue
- Implement the feature or fix the bug
- Open a merge request and describe the changes you made
- Wait for the review
- If the review is successful, the changes will be merged into the main branch
Authors and acknowledgment
A big thanks to the whole iGEM Team Münster 2024 for investing in the development of this tool.
Special thanks to:
- Emil Beurer (Author)
- Jan Albrecht (Author)
- Michel Westermann (Co Author)
- Lasse Middendorf (Advisor)
License
Creative Commons
Attribution 4.0 International (CC BY 4.0) https://creativecommons.org/licenses/by/4.0/
References
-
Biswas, S. et al. (2021) “Low-N protein engineering with data-efficient deep learning,” Nature Methods, 18(4), pp. 389–396. doi: 10.1038/s41592-021-01100-y ↩ ↩2 ↩3
-
Meier, J. et al. (2021) “Language models enable zero-shot prediction of the effects of mutations on protein function.” doi: 10.1101/2021.07.09.450648 ↩ ↩2
-
Milligan, L. et al. (2016) “Strand‐specific, high‐resolution mapping of modified RNA polymerase II,” Molecular Systems Biology, 12(6). doi: 10.15252/msb.20166869 ↩
-
Behrendorff, J. B. Y. H., Johnston, W. A. and Gillam, E. M. J. (2014) “Restriction Enzyme-Mediated DNA Family Shuffling,” in Directed Evolution Library Creation. Springer New York, pp. 175–187. doi: 10.1007/978-1-4939-1053-3_12 ↩ ↩2
-
Johnson, L. S., Eddy, S. R. and Portugaly, E. (2010) “Hidden Markov model speed heuristic and iterative HMM search procedure,” BMC Bioinformatics, 11(1). doi: 10.1186/1471-2105-11-431 ↩
-
Hu, E. J. et al. (2021) “LoRA: Low-Rank Adaptation of Large Language Models.” doi: 10.48550/arxiv.2106.09685 ↩
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file seq_mutator-1.0.0.tar.gz.
File metadata
- Download URL: seq_mutator-1.0.0.tar.gz
- Upload date:
- Size: 61.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.10.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f3232316b6957fd205d91efdbdc04ae939ae31874c51f00dcce8885c761de85f
|
|
| MD5 |
50cc90a75d6bf9c9a5cbb0e97b64841b
|
|
| BLAKE2b-256 |
05bf48de36d64c2012aa8cc7d1198e5d585b70b86bd94f5e1b53aff6bd52abd9
|
File details
Details for the file seq_mutator-1.0.0-py3-none-any.whl.
File metadata
- Download URL: seq_mutator-1.0.0-py3-none-any.whl
- Upload date:
- Size: 55.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.10.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
968cf07516fc5c730bd2bfe3b2ea12d307557e9ecf1c1ad5d787b878e9ab4d7f
|
|
| MD5 |
7e3c32247130725bff3e7ee3de9289a5
|
|
| BLAKE2b-256 |
eaf5d7431dfe07806ed3ecf363da97e0819c719318dafe298f559173745c56d4
|







