This package contains the programs that design primer set for microbiological analysis and perform some accessory analysis.

Project description
Project description
shrs
Primer design method based on short length homologous region exhaustive search algorithm.
Table of Contents
shrs- Table of Contents
- Overview
- Environment
- Installation
- pip
- Installing from the source code
- Download source code
- Installation from source
- How to run
shrs - Functions contained in library of
shrs - Citation
- Update information
- Reference
- License
Overview
This package contains the programs that design a primer set for the analysis of bacteria and perform some accessory analyses. This package contains the following six subcommands.
AA(For Additional analysis)DIP(For design identification primer sets)DUP(For design universal primer sets)ISP(For input sequence preprocessing)iPCR(For insilico PCR)DP(For design probes)
You can get a summary of the available command-line options of each command by using the following command:
shrs, shrs {subcommand} -h.
Every program accepts FASTA or GenBank format sequence files.
The input file must be encoded in UTF-8 format.
Environment
This package passed the operation verification under Windows 10, Windows 11, macOS Sequoia, Ubuntu 18.04, Ubuntu 20.04 and Ubuntu 22.04.
When the 'cupy' package is available, some calculations will be performed using the GPU.
The 'cupy' package is NOT installed automatically, even if you use the pip install shrs command.
Please install the version of the 'cupy' package that matches the version of CUDA on your computer by yourself.
Type pip install shrs[GPU] if you would like to install 'cupy' packages automatically.
Analyses by a PC with a GPU are highly recommended. The processing time when a GPU is used could be less than that when a CPU is used.
Installation
pip
Type the following command.
$ pip install shrs
or
$ pip install shrs[GPU]
Please check above. (Environment section)
Installing from the source code
Download source code
Download the source from the following URL. https://pypi.org/project/shrs/#files
Installation from source
Place the source file on the current working directory, and then type the following commands.
$ tar -xvzf shrs-0.13.2.tar.gz
$ cd shrs-0.13.2
$ python setup.py build
$ python setup.py install
How to run shrs
To confirm a subcommand, type shrs. The following message will be shown.
usage: shrs [-h] [-v] {AA,DIP,DUP,ISP,iPCR,DP} ...
--- HELP message ---
positional arguments:
{AA,DIP,DUP,ISP,iPCR,DP}
AA see 'AA -h'. Make a fragment size matrix from a new template sequence and the result containing primer sets that you have already generated.
DIP see 'DIP -h'. Primer design algorithm for the identification of bacteria
DUP see 'DUP -h'. Primer design algorithm for universal primer
ISP see 'ISP -h'. Input sequences preprocessing program for DUP or DIP
iPCR see 'iPCR -h'. In silico PCR amplification algorithm
DP see 'DP -h'. Probe design algorithm
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit
Sample data
Sample data can be downloaded from here
| Algorithm | Hash value |
|---|---|
| MD5 | 9b40802495ce31bec85b5f541bd6a9d0 |
| SHA1 | ac84bc66e35c126f929d73c444846bec75d75920 |
| SHA256 | da69337717cc4bc101e3474e71b1884d6ba42b8f06772d113f30ff3664cb857e |
Sample data contain a multi-FASTA file of complete genome DNAs of three strains of Mycoplasma genitalium, a FASTA file of the complete genome DNA of one strain of Mycoplasma pneumoniae, a multi-FASTA file of 25 contig sequences of Mycoplasma genitalium G37 and GenBank files of the complete genome DNAs of five strains of Mycoplasma genitalium. Place SampleData in the current working directory.
Workflow for design primer
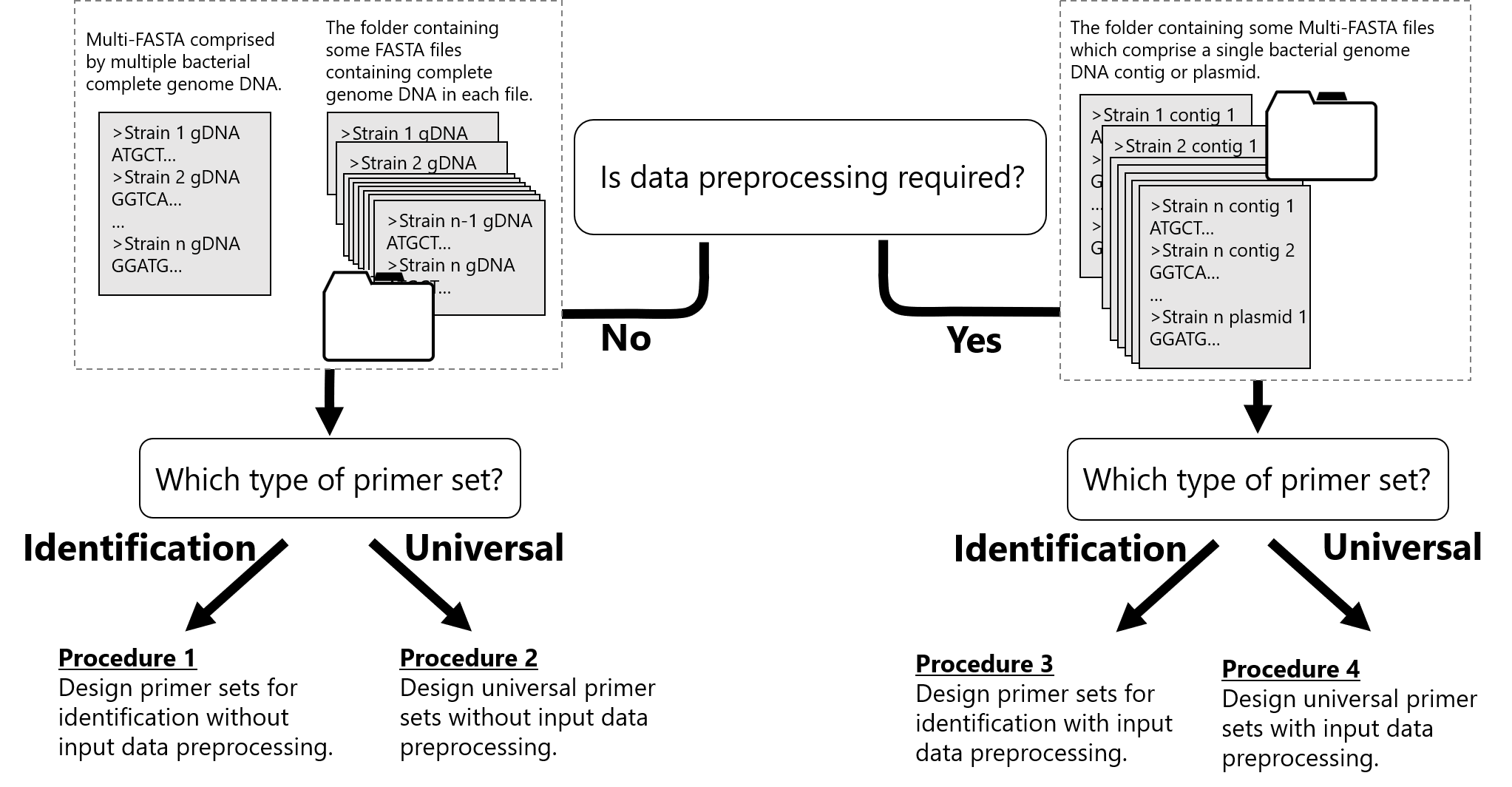
1. Design primer sets for identification without input data preprocessing.
Specify the multi-FASTA file or the folder containing target sequences (FASTA or GenBank format) as input file(s) after the '-i' option.
$ shrs DIP -i SampleData/Mgenitalium_3strains.fasta -o SampleData/ --Search_mode sparse
It takes approx. 20 minutes to 2 hours, depending on the PC's performance. After the analysis, the results will be output in a SampleData/Result/(Timestamp)identify_primer_set/ folder. The CSV file contains a results table as shown below with the analysis parameters.
| Arguments | |
|---|---|
| Input_file_name | sample.fasta |
| Target | Strain1, Strain2, ... , Strain N |
| Exclude_file_name | None |
| Exclude | |
| Probe_size_range_description | 23-25 |
| allowance | 0.15 |
| cut_off_lower | 50 |
| cut_off_upper | 1000 |
| Interval_distance | 10000 |
| Match_rate | 0.8 |
| Result_output | 10000 |
| Search_mode | sparse |
| Window_size | 950 |
| Maximum_annealing_site_number | 5 |
| Score_calculation_mode | Fragment |
| Search_area | Whole genome sequence |
| Reference_tree | Not provided |
| Primer1 | Primer2 | No. | Input sequence1 | Input sequence2 | ... | Input sequenceN | Score | Fragment number | Forward Tm_value | Reverse Tm_value | Tm_value difference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATG..AA | AGC..TA | 1 | [(655, 1.0)] | [(528, 1.0)] | ... | [(411, 1.0)] | 1200 | N | 60.0 | 61.5 | 1.5 |
| TAG..TC | GCC..TG | 2 | [(344, 1.0)] | [(257, 1.0)] | ... | [(499, 1.0)] | 1200 | N | 58.0 | 60.5 | 2.5 |
Input sequence columns indicate the amplicon length amplified by Primers 1 and 2 and its ratio to the total amplicon number [(Amplicon length, Ratio)]. Therefore, if a primer set produces three amplicons (355 bp, 355 bp, and 652 bp) from whole genome DNA, the results would be [(355, 0.67), (652, 0.33)]. Each primer set can be used for identification; however, combining primer sets will make the identification easier and more accurate. When the analysis is run with default settings, three primer sets are selected, and a dendrogram based on the information of fragments amplified by each primer set will be generated in the 'Dendrogram' folder. The 'Combination_number' option (default: 3) can be used to adjust the number of primer sets used for identification. This algorithm DIP designs primer sets that produce fragments from all input sequences and maximizes the differences in the amplicon size or amplicon sequence among input sequences.
To confirm command-line options, type shrs DIP -h.
2. Design universal primer sets without input data preprocessing.
Specify the Multi-FASTA or the folder containing the target sequences (FASTA or GenBank format) as input file(s) after the '-i' option. In the following case, the design primer set can amplify genome DNAs of three strains of Mycoplasma genitalium and NOT produce any amplicons from genome DNA of one strain of Mycoplasma pneumoniae.
$ shrs DUP -i SampleData/Mgenitalium_3strains.fasta -e SampleData/Mpneumoniae.fasta -a 0.25 -o SampleData/ --Search_mode sparse
It takes approx. 5–10 minutes, depending on the PC's performance. After the analysis, the results will be output in a SampleData/Result/(Timestamp)universal_primer_set/ folder. The format of the outputted CSV file is almost the same as that obtained for the results of a DIP analysis as mentioned above (see Procedure 1). This algorithm DUP designs primer sets that produce fragments from all input sequences and minimizes the differences in the amplicon size among input sequences. Note that the primer sets are designed to produce only one amplicon from each input sequence when shrs DUP is used.
To confirm command-line options, type shrs DUP -h.
3. Design primer sets for identification with input data preprocessing.
Each sequence that is input into the DIP subcommand at the same time is processed for differentiation from each other. Therefore, if an inputted multi-FASTA file has plasmid or multiple contig sequences of an identical strain, the primer set obtained will produce some amplicons that correspond to inputted contigs or plasmid. To avoid this problem, if genome DNA of an identical strain has been divided into multiple contigs, all contigs and the plasmid should be preprocessed (concatenated) prior to the DIP analysis, using the following subcommand. One multi-FASTA file for preprocessing must contain the contigs and plasmid derived from a single strain. Organize all multi-FASTA files in one folder when there are some multi-FASTA files, and then specify the folder path as the input file path.
$ shrs ISP -i SampleData/contig/ -o SampleData/Preprocessed_data1/ --Single_file
A preprocessed multi-FASTA file will be generated, and then the preprocessed multi-FASTA file is analyzed by shrs DIP.
$ shrs DIP -i SampleData/Preprocessed_data1/ -o SampleData/ --Search_mode sparse
This algorithm DIP designs primer sets that produce fragments from all input sequences and maximizes the differences in the amplicon size or amplicon sequence among input sequences.
To confirm command-line options, type shrs DIP -h or shrs ISP -h.
4. Design universal primer sets with input data preprocessing.
As mentioned above, if genome DNA of an identical strain has been divided into multiple contigs, all contigs and the plasmid should be preprocessed (concatenated) prior to a DUP analysis by using following subcommand. One multi-FASTA file for preprocessing must contain contigs and plasmid derived from a single strain. Organize all multi-FASTA files in one folder when there are some multi-FASTA files, and then specify the folder path as the input file path.
$ shrs ISP -i SampleData/contig/ -o SampleData/Preprocessed_data2/ --Single_file
A preprocessed multi-FASTA file will be generated, and then the preprocessed multi-FASTA file is analyzed by shrs DUP.
$ shrs DUP -i SampleData/Preprocessed_data2 -e SampleData/Mpneumoniae.fasta -a 0.20 -o SampleData/ --Search_mode sparse
This algorithm DUP designs primer sets that produce fragments from all input sequences and minimizes the differences in the amplicon size among input sequences. Note that the primer sets are designed to produce only one amplicon from each input sequence when shrs DUP is used.
To confirm command-line options, type shrs DUP -h or shrs ISP -h.
Re-analysis/Additional analysis
New sequences can be analyzed based on primer sets in a CSV file obtained from shrs DIP or shrs DUP. Delete the rows containing unnecessary primer sets, since an analysis of them would take a long time. Do not include blank rows between the rows containing primer sets.
$ shrs AA -i SampleData/Mgenitalium_M6282.fasta -f SampleData/Previous_DIP_Result.csv -o SampleData/New_result/
Design probes
Specify the multi-FASTA file or the folder containing target sequences (FASTA or GenBank format) as input file(s) after the '-i' option. This algorithm DP designs probes that hybridize to all input sequences. Input sequence columns in result table indicate the annealing positions on each input sequence.
in silico PCR
You can get amplicon information by using in silico PCR.
$ shrs iPCR -i SampleData/GenBank/ -fwd CTACACCCATTTACCCAACAGTATC -rev TCTCATTGGWAACATTCGGTACATC -o SampleData/insilicoPCR_Result/
A CSV file will be output with default settings. If you prefer FASTA over CSV, use the '--fasta' option. When you need to know the primer annealing site positions on a template sequence, use the '--Position_index' option. You can obtain the annotation information of amplicons, when a GenBank file is analyzed by in silico PCR with the '--Annotation' option.
$ shrs iPCR -i SampleData/GenBank/ -fwd CTACACCCATTTACCCAACAGTATC -rev TCTCATTGGWAACATTCGGTACATC -o SampleData/insilicoPCR_annotation_Result/ --Annotation
If there are some primer sets, you can input a filepath of a text file that contains primer sets after -f option instead of typing primer sequences after -fwd and -rev option. The text file contains one primer set in each row and must be written in with utf-8 encoding. You can specify commas, spaces or tab character as a separator in the text file.
Text file example:
Forward_Primer_sequence1,Reverse_Primer_sequence1
Forward_Primer_sequence2,Reverse_Primer_sequence2
Forward_Primer_sequence3,Reverse_Primer_sequence3
...
Forward_Primer_sequenceN,Reverse_Primer_sequenceN
Tips: Some command line options
-
'--circularDNA'/'--exclude_circularDNA' option {AA, DIP, DUP, ISP, iPCR}
When input sequence file(s) contains one or more circular DNA sequence(s), use this option. Specify which sequence is circular DNA by using a subargument. All input sequences are processed as linear DNA in default settings ('n/a'). When all input sequences are circular DNA, use 'all' after the --circularDNA/--exclude_circularDNA option. For each individual sequence, you can specify whether the sequence is circular DNA or not by using 'individually'. You can also specify whether or not a sequence is circular DNA by inputting the file path of text file, as shown below.
Text file example: Sequence_name1 circularDNA Sequence_name2 linearDNA Sequence_name3 linearDNA ... Sequence_nameN circularDNA
$ shrs DIP -i SampleData/Mgenitalium_3strains.fasta -o SampleData/ --Search_mode sparse --circularDNA SampleData/circularDNA.txt
-
'--Search_mode' option {DIP, DUP}
The '--Search_mode' option contains four choices: 'exhaustive', 'moderate', 'sparse', and 'manual'. Primer candidates will be generated by cutting every N-base from the reference sequence. The N-value is different depending on each Search_mode.
Subargument N-bases exhaustive 1 base. Same as N-gram method (N: primer size) moderate One-third of primer size (Maximum: 10 bases) sparse primer size manual primer size * Ratio inputed by user
$ shrs DIP -i SampleData/Mgenitalium_3strains.fasta -o SampleData/ --Search_mode manual 2
-
'-a' option {DIP, DUP}
When the '-e' and '--Exclude_mode fast' options are used, the larger the value allowance is, the fewer in number the candidates will be.
-
'--Score_calculation' option {DIP}
When 'Sequence' is selected in this option, the cumulative score of sequence homology among amplicons is used as an indicator to evaluate primer set candidates instead of the score calculated from fragment length. When this option is used, only primer sets that produce a single amplicon from the template sequence will be obtained because calculation of a sequence homology takes a long time. Additionally, a multiple alignment tool (MAFFT program) is required to generate a dendrogram when 'Sequence' is specified in 'Score_calculation' option. Before running this script, please install MAFFT and add the binary of MAFFT to your PATH. The dendrogram is constructed based on the UPGMA method, and the distance matrix calculated by the identity matrix. The gap(s) in the alignment(s) are used to calculate distance (NOT ignored). Note that the dendrogram obtained indicates the similarity of alignment sequences containing gap(s), and it does not always indicate the evolutionary relationships.
$ shrs DIP -i SampleData/Mgenitalium_3strains.fasta -o SampleData/ --Search_mode sparse --Score_calculation Sequence
-
'-g' or '--Group_id' option {DIP}
When this option is used, the algorithm tries to minimize the score between the sequences in intra-group and maximize the score between the sequences in inter-group. You can specify which sequences are same group by inputting the file path of text file, as shown below. You can specify commas, spaces or tab character as a separator in the text file. Please avoid to contain a sequence name in ID like as the combination of sequence name 'D12' and ID 'SeqID12'.
Text file example: Sequence_name1 1 Sequence_name2 2 Sequence_name3 3 ... Sequence_nameN 2
$ shrs DIP -i SampleData/Mgenitalium_3strains.fasta -o SampleData/ --Search_mode sparse --Score_calculation Sequence --Group_id SampleData/Group_id.txt
-
'--Fragment_size_pattern_matrix' and/or '--Fragment_start_position_matrix' option {DIP}
When you would like to switch 'Score_calculation mode' to another one (or sometimes generate new dendrograms), it is possible to reanalyze same dataset in less time than before by specifying Fragment_size_pattern_matrix.csv and/or Fragment_start_position_matrix that has already obtained after --Fragment_size_pattern_matrix and/or --Fragment_start_position_matrix option. When 'Fragment' mode will be used, you specify only 'Fragment_size_pattern_matrix.csv'. On the other hand, both 'Fragment_size_pattern_matrix.csv' and 'Fragment_start_position_matrix.csv' have to be specified, when 'Sequence' mode will be used.
-
'--Reference_tree' option {DIP}
Using '--Reference_tree' option, this tool calculates the Robinson-Foulds distance between each generated dendrogram and the reference tree, then sorts the results based on the similarity. When you have a reliable phylogenetic tree, this option makes it easy to select the primer set. The reference tree should be provided as either a character or a file path containing the phylogenetic tree in Newick format following the '--Reference_tree' option. Please note that all leaf names must correspond to the titles of the input sequences. To make things easier to understand, the titles of the sequences in the GenBank file have been changed from original titles in this tutorial.
$ shrs DIP -i SampleData/GenBank/ -o SampleData/ --Search_mode sparse --Reference_tree "(((G37,M6320),M2321),(M2288,M6282));"
-
'--Only_sequence_with_feature_key' option {DIP}
Using '--Only_sequence_with_feature_key' option, this tool generates the primer set based on sorely regions that are annotated with a feature key. At least one GenBank-format file should be used as an input file, as the information about the feature key is required in order to extract the target regions. When this option is specified, all GenBank-format input files are trimmed based on the feature key information. However, Fasta-format files are not trimmed. Note that it does not matter if GenBank and Fasta format files are intermingled in the input sequences, because this tool generates primer sets that are able to amplify all input sequences.
$ shrs DIP -i SampleData/GenBank/ -o SampleData/ --Search_mode sparse --Only_sequence_with_feature_key
Command-line options
Design identification primer sets {DIP}
shrs DIP -i <input file> [options]
Below is the full list of supported options for the DIP command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -e, --exclude_file | File path for exclusion sequence(s). Specify the sequence(s) file path of the bacteria if there are some bacteria that you would like to not amplify. (format: FASTA or Genbank) |
| -s, --primer_size | Primer size (default: 25) |
| -a, --allowance | Mismatch allowance ratio (default: 0.15. The value means that a 4-base [25 * 0.15] mismatch is accepted). Note that setting this parameter too large might causes the increased run time and excessive memory consumption. |
| -r, --range | Search range from primer size (default: 0. If the value is 1, the primer sets that have 25–26 base length are explored) |
| -d, --distance | The minimum distance between annealing sites that are hybridized with a primer (default: 10,000) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| -P, --process | The number of processes (sometimes the number of CPU core) used for analysis |
| -g, --Group_id | Type the file path of the text that specifies which sequences are same group, after the '--Group_id' option. Please avoid to contain a sequence name in ID like as the combination of sequence name 'D12' and ID 'SeqID12'. (default: None.) |
| --Exclude_mode | Choose the method for excluding the sequence(s) from 'fast' or 'standard' when you specify some sequence(s) file path of the bacteria that you would like to exclude using '-e' option (default: fast.) |
| --Result_output | The upper limit of result output (default: 10,000) |
| --Cut_off_lower | The lower limit of amplicon size (default: 50) |
| --Cut_off_upper | The upper limit of amplicon size (default: 1,000) |
| --Match_rate | The ratio of trials for which the primer candidate fulfilled all criteria when the allowance value is decreased. The higher the number is, the more specific the primer obtained is (default: 0.8) |
| --Chunks | The chunk size for calculation. If the memory usage for calculation using the chunk size exceeds the GPU memory size, the processing unit for calculation will be switched to the CPU automatically (default: Auto) |
| --Maximum_annealing_site_number | The maximum acceptable value of the number of annealing site of the candidate of the primer in the input sequence (default: 5) |
| --Window_size | The duplicated candidates containing this window will be removed (default: 950) |
| --Search_mode | There are four options: exhaustive/moderate/sparse/manual. If you choose the 'manual' option, type the ratio to primer length after 'manual'. (e.g. --Search_mode manual 0.2.) (default: moderate when the average input sequence length is >5000, and exhaustive when the average input sequence length is ≤5000) |
| --withinMemory | All analyses are performed within memory (default: False) |
| --Without_allowance_adjustment | Use this option if you do not want to modify the allowance value for every homology calculation (default: False) |
| --circularDNA | If there are some circular DNAs in the input sequences, use this option (default: n/a. It means all input sequences are linear DNA. When there are some circular DNA input sequences, type 'all', 'individually', 'n/a', or the file path of the text that specify which sequence is circularDNA, after the '--circularDNA' option.) |
| --exclude_circularDNA | If there are some circular DNAs in the input sequence(s) that you do not want to amplify, use this option (default: n/a. It means all input sequences are linear DNA. When there is some circular DNA in the sequence file for exclusion, type 'all', 'individually', 'n/a', or the file path of the text that specifies which sequence is circularDNA, after the '--exclude_circularDNA' option.) |
| --Score_calculation | The calculation method of the score for identifying microorganisms. Fragment length or sequence. When the 'Sequence' is specified, the primer set that produces only a single amplicon will be obtained in order to reduce computational complexity. |
| --Combination_number | The number of primer sets to be used for identification (default: 3). |
| --Correlation_threshold | The primer sets with a correlation coefficient greater than this are grouped, and two or more primer sets from the same group are never chosen sets (default: 0.9). |
| --Dendrogram_output | The number supplied in this parameter will be used to construct dendrograms. As a result, the default parameters yield 10 dendrograms (default: 10, max: 100). |
| --Reference_tree | Use this option when evaluating a primer set based on Robinson-Foulds distance between the output and reference trees. After the '--Reference_tree' option, specify either a phylogenetic tree in Newick format or the file path to one. (default: None). |
| --Only_sequence_with_feature_key | This option should be used when designing primer sets based solely on the sequences with a feature key, such as a gene. (default: False). |
| --Fragment_size_pattern_matrix | When you have a csv file of fragment size pattern matrix, you can reanalyse from the csv file. Specify the file path (default: None.) |
| --Fragment_start_position_matrix | When you reanalyse from fragment size pattern matrix by 'Sequence' mode, specify the csv file path of fragment start position matrix (default: None.) |
Design universal primer sets {DUP}
shrs DUP -i <input file> [options]
Below is the full list of supported options for the DUP command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| -e, --exclude_file | File path for exclusion sequence(s). Specify the sequence(s) file path of the bacteria if there are some bacteria that you would like to not amplify. (format: FASTA or Genbank) |
| -s, --primer_size | Primer size (default: 20) |
| -a, --allowance | Mismatch allowance ratio (default: 0.20. The value means that a 4-base [20 * 0.20] mismatch is accepted). Note that setting this parameter too large might causes the increased run time and excessive memory consumption. |
| -r, --range | Search range from primer size (default: 0. If the value is 1, the primer sets that have 20–21 base length are explored) |
| -d, --distance | The minimum distance between annealing sites that are hybridized with a primer (default: 5,000) |
| -P, --process | The number of processes (sometimes the number of CPU core) used for analysis |
| --Exclude_mode | Choose the method for excluding the sequence(s) from 'fast' or 'standard' when you specify some sequence(s) file path of the bacteria that you would like to exclude using '-e' option (default: fast.) |
| --Cut_off_lower | The lower limit of amplicon size (default: 50) |
| --Cut_off_upper | The upper limit of amplicon size (default: 3,000) |
| --Match_rate | The ratio of trials for which the primer candidate fulfilled all criteria when the allowance value is decreased. The higher the number is, the more specific the primer obtained is (default: 0.0) |
| --Result_output | The upper limit of result output (default: 10,000) |
| --Omit_similar_fragment_size_pair | Use this option if you want to omit primer sets that amplify similar fragment lengths |
| --Window_size | The duplicated candidates containing this window will be removed (default: 50) |
| --Maximum_annealing_site_number | The maximum acceptable value of the number of annealing site of the candidate of the primer in the input sequence (default: unlimited) |
| --Chunks | The chunk size for calculation. If the memory usage for calculation using the chunk size exceeds the GPU memory size, the processing unit for calculation will be switched to the CPU automatically (default: Auto) |
| --Search_mode | There are four options: exhaustive/moderate/sparse/manual. If you choose the 'manual' option, type the ratio to primer length after 'manual'. (e.g. --Search_mode manual 0.2.) (default: moderate when the average input sequence length is >5000, and exhaustive when the average input sequence length is ≤5000) |
| --withinMemory | All analyses are performed within memory (default: False) |
| --Without_allowance_adjustment | Use this option if you do not want to modify the allowance value for every homology calculation (default: False) |
| --circularDNA | If there are some circular DNAs in the input sequences, use this option (default: n/a. It means all input sequences are linear DNA. When there are some circular DNA input sequences, type 'all', 'individually', 'n/a', or the file path of the text that specify which sequence is circularDNA, after the '--circularDNA' option.) |
| --exclude_circularDNA | If there are some circular DNAs in the input sequence(s) that you do not want to amplify, use this option (default: n/a. It means all input sequences are linear DNA. When there is some circular DNA in the sequence file for exclusion, type 'all', 'individually', 'n/a', or the file path of the text that specifies which sequence is circularDNA, after the '--exclude_circularDNA' option.) |
| --Fragment_size_pattern_matrix | When you have a csv file of fragment size pattern matrix, you can reanalyse from the csv file. Specify the file path (default: None.) |
Input Sequence Preprocessing {ISP}
shrs ISP -i <input file> [options]
Below is the full list of supported options for the ISP command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| --circularDNA | If there are some circular DNAs in the input sequences, use this option (default: n/a. It means all input sequences are linear DNA. When there are some circular DNA input sequences, type 'all', 'individually', 'n/a', or the file path of the text that specify which sequence is circularDNA, after the '--circularDNA' option.) |
| --circularDNAoverlap | Maximum value of overlapped region in circular DNA (default: 10,000). For reducing computational complexity, you can reduce this value to the larger one of the upper limit of amplicon size and interval distance that will be set by '--Cut_off_upper' and '--distance' in following DIP or DUP. |
| --Single_target | All input files will be concatenated and generated as one sequence if you use this option, even if separated multi-FASTA files are inputted. |
| --Multiple_targets | All input files are recognized as an individual file, and every file is preprocessed separately. |
| --Single_file | When you use the '--Multiple_targets' option and this option, a preprocessed sequence file will be outputted as one multi-FASTA file. |
Additional Analysis {AA}
shrs AA -i <input file> -f <csv file> [options]
Below is the full list of supported options for the AA command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -f, --csv_file | File path of the CSV data obtained from other analysis (DIP or DUP) (required) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| -s, --size_limit | The upper limit of amplicon size (default: 3,000) |
| -P, --process | The number of processes (sometimes the number of CPU core) used for analysis |
| -fwd, --forward | Forward primer sequence (required if you don't provide a CSV file with the '-f' option) |
| -rev, --reverse | Reverse primer sequence (required if you don't provide a CSV file with the '-f' option) |
| --circularDNA | If there are some circular DNAs in the input sequences, use this option (default: n/a. It means all input sequences are linear DNA. When there are some circular DNA input sequences, type 'all', 'individually', 'n/a', or the file path of the text that specify which sequence is circularDNA, after the '--circularDNA' option.) |
| --warning | Shows all warnings when you use this option |
in silico PCR {iPCR}
shrs iPCR -i <input file> -fwd <forward primer sequence> -rev <reverse primer sequence> [options]
Below is the full list of supported options for the iPCR command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| -s, --size_limit | The upper limit of amplicon size (default: 10,000) |
| -P, --process | The number of processes (sometimes the number of CPU core) used for analysis |
| -fwd, --forward | The forward primer sequence used for amplification (required) |
| -rev, --reverse | The reverse primer sequence used for amplification (required) |
| -f, --primerset_filepath | The filepath of the text file containing forward and reverse primer sequence (required if forward and reverse primer set with -fwd and -rev option are not provided) |
| --fasta | Output format. A FASTA file will be generated if you use this option. |
| --Single_file | Output format. One single FASTA-format file will be generated even if you input some separate FASTA files, when using this option with the '--fasta' option. |
| --Mismatch_allowance | The acceptable mismatch number (default: 0) |
| --Only_one_amplicon | Only one amplicon is outputted, even if multiple amplicons are obtained by PCR when you use this option. |
| --Position_index | The result has the information of the amplification position when this option is enabled. |
| --circularDNA | If there are some circular DNAs in the input sequences, use this option (default: n/a. It means all input sequences are linear DNA. When there are some circular DNA input sequences, type 'all', 'individually', 'n/a', or the file path of the text that specify which sequence is circularDNA, after the '--circularDNA' option.) |
| --gene_annotation_search_range | The gene annotation search range in the GenBank-format file. (default: 100) |
| --LowQualitySequences | In in silico PCR analysis, all sequences containing the regions with a high proportion of 'N' bases will be omitted when you specify 'remove' option. This option helps to reduce a computational effort and calculation time. If you select the omitted sequences individually, specify 'individually'. When the 'ignore' option is selected, regions spanning 'N' bases will not be amplified. To use all input sequences for the in silico PCR template, specify the 'remain' option. (Default: 'remain') |
| --Annotation | If the input sequence file is in GenBank format, the amplicon(s) is annotated automatically. |
| --warning | Shows all warnings when you use this option |
Design probes {DP}
shrs DP -i <input file> [options]
Below is the full list of supported options for the DP command line.
| Option | Description |
|---|---|
| -i, --input_file | Input file path (required, format: FASTA or Genbank) |
| -e, --exclude_file | File path for exclusion sequence(s). Specify the sequence(s) file path of the bacteria if there are some bacteria that you would like to not hybridize. (format: FASTA or Genbank) |
| -s, --probe_size | Probe size (default: 25) |
| -a, --allowance | Mismatch allowance ratio (default: 0.25. The value means that a 7-base [25 * 0.25] mismatch is accepted). Note that setting this parameter too large might causes the increased run time and excessive memory consumption. |
| -r, --range | Search range from probe size (default: 0. If the value is 1, the probe that have 25–26 base length is explored) |
| -d, --distance | The minimum distance between annealing sites that are hybridized with a probe (default: 100) |
| -o, --output | Output directory path (Make a new directory if the directory does not exist) |
| -P, --process | The number of processes (sometimes the number of CPU core) used for analysis |
| --Exclude_mode | Choose the method for excluding the sequence(s) from 'fast' or 'standard' when you specify some sequence(s) file path of the bacteria that you would like to exclude using '-e' option (default: fast.) |
| --Result_output | The upper limit of result output (default: 10,000) |
| --Match_rate | The ratio of trials for which the probe candidate fulfilled all criteria when the allowance value is decreased. The higher the number is, the more specific the probe obtained is (default: 0.8) |
| --Chunks | The chunk size for calculation. If the memory usage for calculation using the chunk size exceeds the GPU memory size, the processing unit for calculation will be switched to the CPU automatically (default: Auto) |
| --Maximum_annealing_site_number | The maximum acceptable value of the number of annealing site of the candidate of the probe in the input sequence (default: 5) |
| --Search_mode | There are four options: exhaustive/moderate/sparse/manual. If you choose the 'manual' option, type the ratio to probe length after 'manual'. (e.g. --Search_mode manual 0.2.) (default: moderate when the average input sequence length is >5000, and exhaustive when the average input sequence length is ≤5000) |
| --withinMemory | All analyses are performed within memory (default: False) |
| --Without_allowance_adjustment | Use this option if you do not want to modify the allowance value for every homology calculation (default: False) |
Functions contained in library of shrs
- shrslib.basicfunc
- class nucleotide_sequence
- complementary_sequence
- calculate_Tm_value
- read_sequence_file
- shrslib.explore
- search_position
- PCR_amplicon
- shrslib.scores
- calculate_flexibility
- calculate_score
- calculate_diff_length_score
- fragment_size_distance
- array_diff
- sequence_duplicated
See documentation for more detailed information.
Citing SHRS
Please cite the following article:
Takahashi, M., Morikawa, K., Akao, T., 2022. Short-length Homologous Region exhaustive Search algorithm (SHRS): A primer design algorithm for differentiating bacteria at the species, subspecies, or strain level based on a whole genome sequence. J. Microbiol. Methods 203, 106605. DOI: 10.1016/j.mimet.2022.106605
Update information
Version 0.10.0: The argument '--Group_id' has been added to DIP mode.
Version 0.11.0: New ability for designing a probe has been added.
Version 0.12.0: The argument '-f, --primerset_filepath' has been added to iPCR mode.
Version 0.13.0: The '--Reference_tree' and '--Only_sequence_with_feature_key' arguments have been added to the DIP mode. The '--Reference_tree' option sorts the results by Robinson-Foulds distance in comparison to the provided reference tree. The '--Only_sequence_with_feature_key' option generates primer sets based on regions annotated with a feature key (For more information, see Tips section above). The default value of the number of processors used in in silico PCR analysis has been changed to '1'.
Reference
The sequences used in this tutorial are below. All sequences have been downloaded from INSDC member database and RefSeq database.
| Strain name | Accession | Format |
|---|---|---|
| Mycoplasma genitalium G37 | L43967 | GenBank |
| Mycoplasma genitalium G37 | AAGX01000001 - AAGX01000025 | Fasta |
| Mycoplasma genitalium M2288 | CP003773 | GenBank |
| Mycoplasma genitalium M2321 | CP003770 | GenBank & Fasta |
| Mycoplasma genitalium M6282 | NC_018496 | GenBank & Fasta |
| Mycoplasma genitalium M6320 | CP003772 | GenBank & Fasta |
| Mycoplasma pneumoniae NCTC10119 | NZ_LR214945 | Fasta |
License
This software is released under the MIT License. Copyright (c) 2022 Masayuki TAKAHASHI
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file shrs-0.13.2.tar.gz.
File metadata
- Download URL: shrs-0.13.2.tar.gz
- Upload date:
- Size: 112.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9bde663c76bb986365a75533920e8268dea7f8e80578fa2d8bab878c2646c567
|
|
| MD5 |
bf28dbec529ed1f53106dbf2d77c60ca
|
|
| BLAKE2b-256 |
ea062f5f51eb0c20a806c3bb615cfd943396a008f15b187e5dee0ecf4478af27
|
File details
Details for the file shrs-0.13.2-py3-none-any.whl.
File metadata
- Download URL: shrs-0.13.2-py3-none-any.whl
- Upload date:
- Size: 109.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b8afbc3d31389d3a32d015a0c5d28018dcfa9a41595042b537f229d4b7ca3a83
|
|
| MD5 |
84b6c87914b3858a42635bfa764d60fc
|
|
| BLAKE2b-256 |
86c529e1c41cbac9218db09d6665503ecc123cc9c983f2d77d4adad27e643baf
|







