SimPhyni: a tool for phylogenetic trait simulation and inference.

Project description
SimPhyNI
Overview
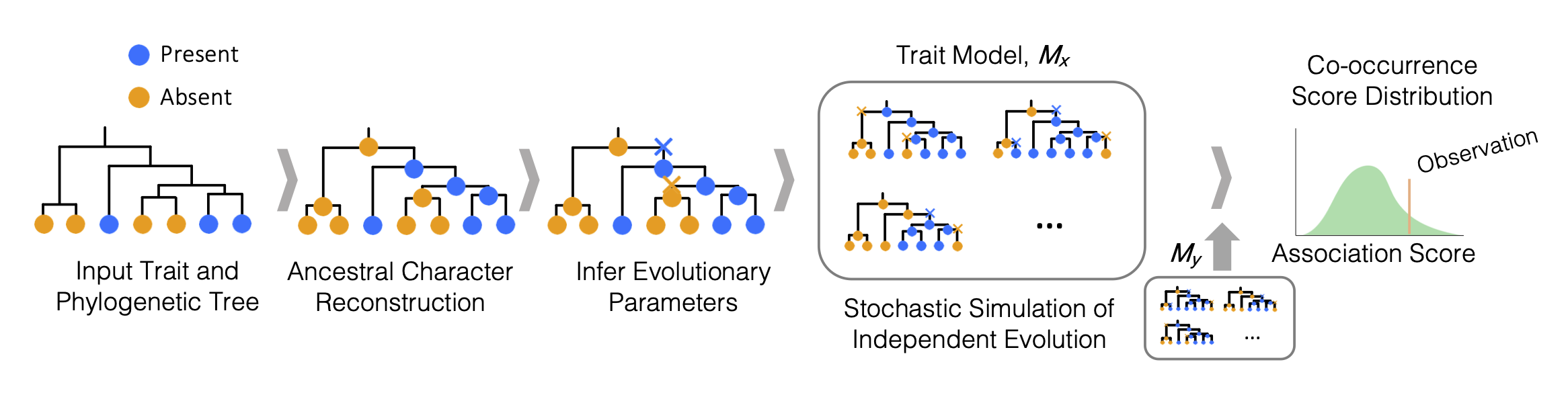
SimPhyNI (Simulation-based Phylogenetic iNteraction Inference) is a phylogenetically-aware framework for detecting evolutionary associations between binary traits (e.g., gene presence/absence, major/minor alleles, binary phenotypes) on microbial phylogenetic trees. This tool leverages phylogenetic information to correct for spurious associations caused by the relatedness of sister taxa.
This pipeline is designed to:
- Infer evolutionary parameters for traits (gain/loss rates, time to emergence, ancestral states)
- Estimate trait co-occurrence null models through independent simulation of traits
- Output statistical results for associations
Graphical Workflow:
Getting Started
Installation
First, ensure bioconda and conda-forge channels are configured:
conda config --add channels conda-forge
conda config --add channels bioconda
Create a new environment:
conda create -n simphyni
conda activate simphyni
Then install SimPhyNI from bioconda:
conda install simphyni
Test installation:
simphyni version
Input Specifications
1. Phylogenetic Tree (.nwk)
- Standard Newick format.
- Must be rooted (both outgroup and midpoint are acceptable).
- Tip labels must match the sample name index in your traits file.
- Branch lengths are required for accurate rate estimation.
2. Traits File (.csv or .parquet)
- Rows: Genomes/Samples (matching tree tips).
- Columns: Binary traits (0 = Absent, 1 = Present; non-numerical values will be set to 1 and blank values will be set to 0).
- Header: Required (Trait names).
- Index: The first column must contain sample names matching tree tip labels.
For large datasets (>10,000 traits), Parquet format is strongly recommended. Parquet enables column-projection and row-predicate pushdown, so only the data actually needed is loaded from disk, dramatically reducing peak memory use.
Example traits.csv:
Sample,PhenotypeX,GeneA,GeneB
E_coli_1,1,0,1
E_coli_2,1,1,0
E_coli_3,0,0,1
Starting from FASTA files and looking for gene-gene or gene-phenotype associations?
If you have raw genome assemblies (FASTA) and need to generate the necessary inputs (gene presence/absence and a phylogenetic tree), we provide a dedicated pipeline: SimPhyNI-Prelude.
This Snakemake workflow is configured for HPC and automates the following steps:
- Annotation (Prokka)
- Pangenome Analysis (Panaroo)
- Tree Construction (PopPUNK or RAxML)
- Formatting (Preparation for SimPhyNI)
- SimPhyNI Analysis (This repository)
Any steps may be bypassed by providing existing data (e.g. gene annotations, phylogenetic tree).
For those familiar with Snakemake, rules can be edited, added, or removed to suit your needs.
Usage
Run mode (single-run)
simphyni run \
--sample-name my_sample \
--tree path/to/tree.nwk \
--traits path/to/traits.csv \
--run-traits 0,1,2 \
--outdir my_analysis \
--cores 4 \
--temp-dir ./tmp \
--min_prev 0.05 \
--max_prev 0.95 \
--plot
Key flags:
--run-traits— Comma-separated list of N values (e.g.0,1,2) to designate the first N traits in the file as query traits for a one-vs-rest comparison. UseALL(default) for all-against-all. For example,--run-traits 0,1,2tests the first 3 traits against all others.--include-flagged— By default, traits whose Poisson null distributions are detected as miscalibrated (sparse events combined with highly asymmetric eligible regions) are excluded from testing and assigned p=0.5. Pass--include-flaggedto simulate these traits anyway; their results will be marked withnull_calibrated=Falsein the output so you can assess them separately. Access the list of flagged traits viaSim.get_flagged_traits()when using the Python API.--min_prev/--max_prev— Minimum and maximum tip prevalence thresholds for traits to be included (default: 0.05 / 0.95).--plot— Generate heatmap summaries of results.--save-object— Save the full analysis object as a.pklfile (not recommended for analyses > 1,000,000 trait pairs).
Run mode (batch)
Create a samples.csv file:
Sample,Tree,Traits,run_traits,MinPrev,MaxPrev
run1,tree1.nwk,traits1.csv,ALL,0.05,0.95
run2,tree2.nwk,traits2.csv,"0,1,2",0.05,0.90
run_traits,MinPrev, andMaxPrevare optional columns that will use default values if not provided.run_traitsmust beALL(case-sensitive) or a comma-separated list of N values (e.g.0,1,2to query the first 3 traits against all others).
Then execute:
simphyni run --samples samples.csv --cores 16
Run with HPC
First, download the cluster profile template:
simphyni download-cluster-profile
Edit cluster_profile/config.yaml for your computing cluster, then install the appropriate Snakemake executor from the available catalog: https://snakemake.github.io/snakemake-plugin-catalog/index.html (SLURM shown below):
pip install snakemake-executor-plugin-slurm
Run SimPhyNI with the --profile flag:
simphyni run --samples samples.csv --profile cluster_profile
For all run options:
simphyni run --help
Example data
Download and run example inputs using:
simphyni download-examples
simphyni run --samples example_inputs/simphyni_sample_info.csv --cores 8 --plot
Outputs
Outputs for each sample are placed in structured folders in the working directory or specified output directory in subdirectories by sample name, including:
Main Result Files
simphyni_results.csv
Contains the statistical results for all tested trait pairs.
| Column | Description |
|---|---|
T1 / T2 |
Identifiers for the two traits being compared. |
direction |
Direction of association: 1 = Positive, -1 = Negative. |
effect size |
Variance-adjusted magnitude of the association. |
prevalence_T1 / prevalence_T2 |
Fraction of samples containing each trait (0.0 to 1.0). |
pval_naive |
Raw empirical P-value from the simulation. |
pval_bh |
Benjamini-Hochberg FDR correction (recommended for phenotype-genotype tests). |
pval_by |
Benjamini-Yekutieli FDR correction (recommended for genotype-genotype tests; accounts for correlated hypotheses, which is the typical case for phylogenetically structured data). |
pval_bonf |
Bonferroni correction (strictest; use when a small number of specific hypotheses are tested). |
null_calibrated |
True for standard results. False for pairs involving traits whose null distributions were detected as miscalibrated (only present when --include-flagged is used). |
Additional Outputs
simphyni_results.csv: Main results table (see above).simphyni_object.pkl: Optional file containing the completed analysis object, parsable with an active SimPhyNI environment. Controlled with the--save-objectflag (not recommended for large analyses > 1,000,000 comparisons).- Plots: Heatmap summaries of tested associations (if
--plotis enabled).
Directory Structure
SimPhyNI/
├── simphyni/ # Core package
│ ├── Simulation/ # Simulation engine
│ ├── scripts/ # Pipeline scripts
│ ├── Snakefile.py # Workflow definition
│ ├── simphyni_cli.py # Command-line entry points
│ └── envs/simphyni.yaml # Conda environment (used by Snakemake)
├── tests/ # Testing suite
├── conda-recipe/ # Build recipe
├── cluster_profile/ # Cluster config template for HPC
├── example_inputs/ # Example inputs to run SimPhyNI
└── pyproject.toml
Contact
For questions, please open an issue or contact Ishaq Balogun at https://github.com/jpeyemi.
Citation
If you use SimPhyNI in your research, please cite:
High Precision Binary Trait Association on Phylogenetic Trees Ishaq O Balogun, Christopher P Mancuso, Tami D Lieberman bioRxiv 2025.12.24.696407; doi: https://doi.org/10.64898/2025.12.24.696407
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file simphyni-2.0.0.tar.gz.
File metadata
- Download URL: simphyni-2.0.0.tar.gz
- Upload date:
- Size: 107.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
0df1b5f2ad3e6e8538d61f4f61e74344775c79e1a70e5452e2f7e567f67fac02
|
|
| MD5 |
9ccff87432744866a6f917f95f5b3136
|
|
| BLAKE2b-256 |
4ce91746062aae3609617388869b59e8dcd28b83e430794fe0a0ae822d5730ae
|
File details
Details for the file simphyni-2.0.0-py3-none-any.whl.
File metadata
- Download URL: simphyni-2.0.0-py3-none-any.whl
- Upload date:
- Size: 76.7 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
76b791cfd3f679ebf6dfdcf5d0ce419378aa6a2ec048fe2faf22014af9241cef
|
|
| MD5 |
5574521f1fa1c3e5d96c612c9c639009
|
|
| BLAKE2b-256 |
82cc643a0922e4a661aa8541d2a04bfe82aa555b57c3d972c3b24656f6ff0a54
|







