







Teloclip
A tool for the recovery of unassembled telomeres from raw long-reads using soft-clipped read alignments.
🎉🧬 Teloclip supports automatic telomere extension with teloclip extend!! 🧬🎉
📖 Full documentation — tutorial, guidance on interpreting results, and CLI reference.
Table of contents
- About Teloclip
- CLI Structure
- Options and Usage
- Example Usage
- Options
- Citing Teloclip
- Publications using Teloclip
- Issues
- License
About Teloclip
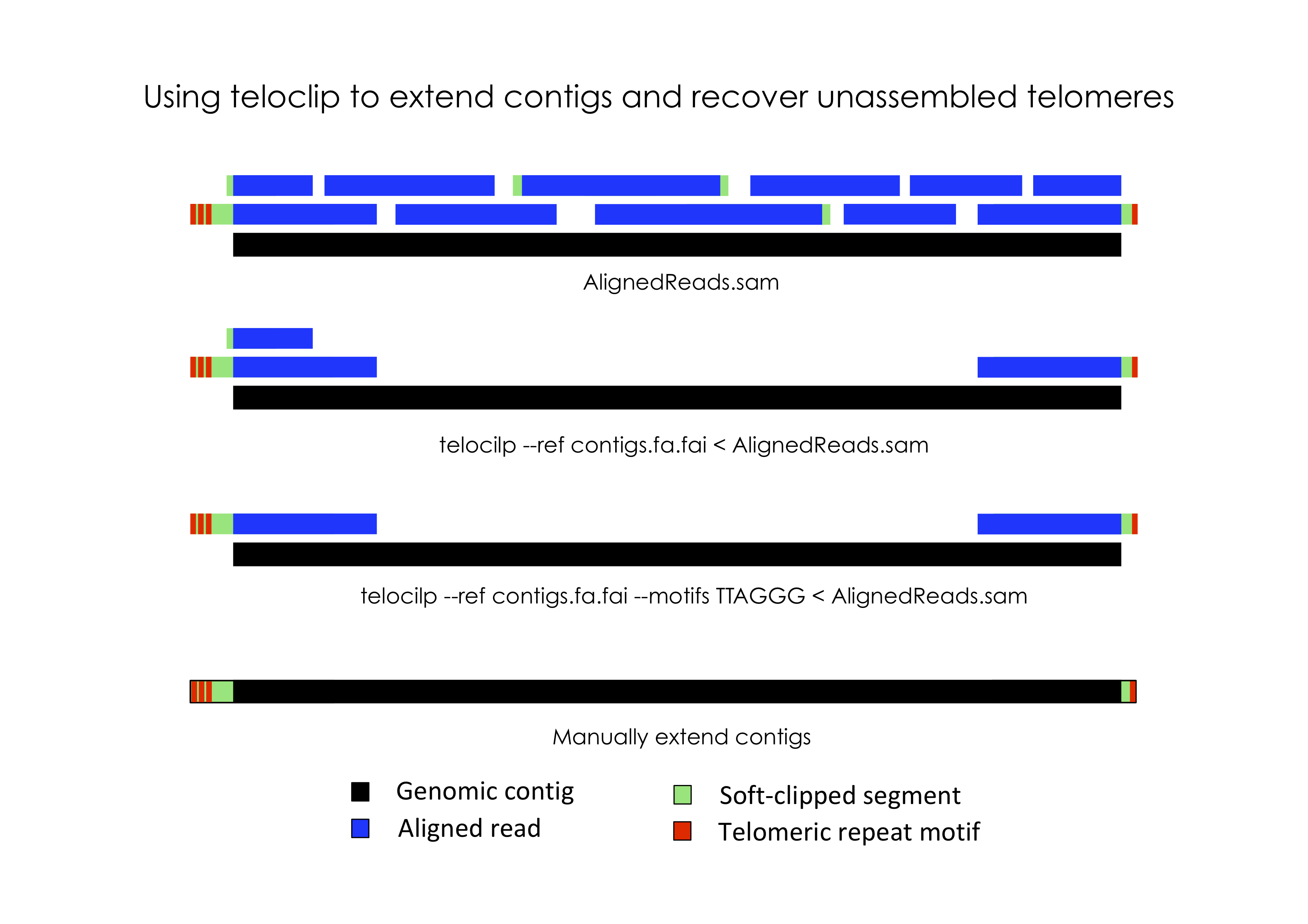
In most eukaryotic species, chromosomes terminate in repetitive telomeric sequences. A complete genome assembly should ideally comprise chromosome-level contigs that possess telomeric repeats at each end. However, genome assemblers frequently fail to recover these repetitive features, instead producing contigs that terminate immediately prior to telomeric repeats.
Teloclip is designed to scan raw long-read data for evidence that can be used to restore missing telomeres. It does this by searching alignments of raw long-read data (i.e. Pacbio or ONT reads mapped with Minimap2) for 'clipped' alignments that occur at the ends of draft contigs. A 'clipped' alignment is produced where the end of a read is not part of its best alignment. This can occur when a read extends past the end of an assembled contig.
Information about segments of a read that were aligned or clipped are stored in SAM formatted alignments as a CIGAR string. Teloclip parses these strings to determine if a read has been clipped at one or both ends of a contig.
Optionally, teloclip can screen overhanging reads for telomere-associated motifs (i.e. 'TTAGGG' / 'CCCTAA') and report only those containing a match.
Once candidate telomeric sequences have be detected in alignment overhangs, teloclip can be used to automatically patch the missing sequence onto draft contigs.
Teloclip is based on concepts from Torsten Seemann's excellent tool samclip. Samclip can be used to remove clipped alignments from a samfile prior to variant calling.
CLI Structure
Teloclip provides three sub-commands:
teloclip filter: Filter SAM/BAM files to identify terminal soft-clipped alignments containing potential telomeric sequencesteloclip extract: Extract overhanging reads to separate FASTA files organized by contig and end positionteloclip extend: Extend draft contigs using overhang analysis from soft-clipped alignments.
Options and Usage
Installation
Teloclip requires Python >= 3.8.
There are 5 options available for installing Teloclip locally:
- Install from PyPi. This or Bioconda will get you the latest stable release.
pip install teloclip
- Install from Bioconda.
conda install -c bioconda teloclip
- Pip install directly from this git repository.
This is the best way to ensure you have the latest development version.
pip install git+https://github.com/Adamtaranto/teloclip.git
- Use Docker for reproducible containerized environments.
Ideal for pipelines and reproducible workflows. No local Python installation required.
# Pull the latest image
docker pull adamtaranto/teloclip:latest
# Run teloclip
docker run --rm -v $(pwd):/data adamtaranto/teloclip:latest --version
See DOCKER.md for complete Docker usage guide and examples/nextflow/ for Nextflow integration.
Verify installation
# Print version number and exit.
teloclip --version
# > teloclip, version 0.3.5
# Get usage information
teloclip --help
Example Usage
Basic use case:
First index the reference assembly so teloclip knows where each contig ends.
# Create index of reference fasta
samtools faidx ref.fa
Next align your raw long reads to the reference fasta.
minimap2 -ax map-pb ref.fa pacbio_reads.fq.gz > in.sam
Loading alignments from file
Next you will need to provide alignment records to teloclip in SAM format. These can be read directly from a SAM file like this:
# Option 1: Read alignment input from sam file and write overhang-reads to stdout
teloclip filter --ref-idx ref.fa.fai in.sam
# Option 2: Read alignment input from stdin and write stdout to file
teloclip filter --ref-idx ref.fa.fai < in.sam > overhangs.sam
Alternatively, you can read and write alignment records from BAM files.
BAM files are binary SAM files, they contain all the same information but take up much less storage space.
You can use BAM files with teloclip like this:
# Read alignments from bam file, pipe sam lines to teloclip, sort overhang-read alignments and write to bam file
samtools view -h in.bam | teloclip filter --ref-idx ref.fa.fai | samtools sort > overhangs.bam
Streaming alignments from Minimap
You can also stream SAM records directly from the aligner to save disk space.
# Map PacBio long-reads to ref assembly,
# return alignments clipped at contig ends,
# write to sorted bam.
minimap2 -ax map-pb ref.fa pacbio_reads.fq.gz | teloclip filter --ref-idx ref.fa.fai | samtools sort > overhangs.bam
Report clipped alignments containing target motifs
teloclip filter has the option to report only overhanging reads that contain a known telomeric repeat sequence.
# Report alignments which are clipped at a contig end
# AND contain >=1 copy of the telomeric repeat "TTAGGG" (or its reverse complement "CCCTAA") in the clipped region.
samtools view -h in.bam | teloclip filter --ref-idx ref.fa.fai --motifs TTAGGG | samtools sort > overhangs.bam
# To change the minimum number of consecutive motif repeats required for a match, set "--min-repeats". This example will require one instance of "TTAGGGTTAGGGTTAGGG" in the overhang.
samtools view -h in.bam | teloclip filter --ref-idx ref.fa.fai --motifs TTAGGG --min-repeats 3 | samtools sort > out.bam
Matching noisy target motifs
Raw long-reads can contain errors in the length of homopolymer tracks. If the --fuzzy option is set, motifs will be converted to regex patterns that allow the number of repeated bases to vary by +/- 1.
i.e. "TTAGGG" -> "T{1,3}AG{2,4}". This pattern will match TTAGG TTAGGGG TAGG TTTAGGG etc.
To reduce off target matching you can increase the minimum required number of sequential motif matches with "--min-repeats".
samtools view -h in.bam | teloclip filter --ref-idx ref.fa.fai --fuzzy --motifs TTAGGG --min-repeats 4 | samtools sort > overhangs.bam
Extract clipped reads
teloclip extract will write overhanging reads to separate fasta files for each reference contig end. The clipped region of each read is masked as lowercase in output fasta files.
You can inspect these reads and select candidates to manually extend contig ends.
# Find soft-clipped alignments containing motif 'TTAGGG' that overhang contig ends, write to sorted bam.
samtools view -h in.bam | teloclip filter --ref-idx ref.fa.fai --motifs TTAGGG | samtools sort > sorted_overhangs.bam
# Extract overhang reads and write to separate fasta files for each reference contig end.
# Adds overhang stats to fasta header and writes overhang region in lowercase.
# Note: Use sorted input to make processing more efficient.
samtools view -h sorted_overhangs.bam | teloclip extract --ref-idx ref.fa.fai --extract-dir split_overhangs_by_contig --include-stats --count-motifs TTAGGG --report-stats
Automatically extend missing telomeres
Use the teloclip extend tool to automatically extend contigs with missing telomeic sequences from overhang-reads identified with teloclip filter.
Before using overhangs identified by Teloclip to extend contigs you should inspect the alignments in a genome browser that displays information about clipped reads, such as IGV.
Check for conflicting soft-clipped sequences. These indicate non-specific read alignments. You may need to tighten your alignment criteria or manually remove low-confidence alignments.
Note: Circular genomes (i.e. mitochondria, chloroplasts, and nitroplasts) will always yield soft-clipped overhangs and should not be extended. Exclude them by name with --exclude-contigs (or list them one per line and pass --exclude-contigs-file).
Teloclip also reports contigs whose overhang coverage is far above the rest of the assembly, which is the usual signature of a collapsed repeat, an rDNA array, or an organellar contig attracting reads from elsewhere. These appear in the stats report under Contigs With Anomalous Overhang Coverage. They are not excluded automatically: whether extension is appropriate is a judgement about your assembly. Review them and re-run with --exclude-contigs if you agree.
--exclude-outliersis deprecated and now does nothing. It previously dropped such contigs silently, and the exclusions were never reported anywhere.
# Create required indices (one-time setup)
samtools faidx ref.fa
# Convert SAM -> BAM, sort, and write sorted BAM
samtools view -bS overhangs.sam | samtools sort -o overhangs.sorted.bam
# Index the sorted BAM for fast access
samtools index overhangs.sorted.bam
# Use `--dry-run` to report proposed changes without applying them.
teloclip extend overhangs.sorted.bam ref.fa \
--output-fasta extended.fasta \
--stats-report extension_report.md \
--count-motifs TTAGGG \
--screen-terminal-bases 1000 \
--exclude-contigs ctg_007_mitochondrial \
--dry-run
# Record every overhang read that was considered, and keep a run log.
teloclip extend overhangs.sorted.bam ref.fa \
--output-fasta extended.fasta \
--stats-report extension_report.md \
--overhang-log overhangs.tsv \
--logfile teloclip_extend.log
--stats-report accepts a path, or - to write the report to stdout. If
omitted it goes to stderr, interleaved with the log.
Extension amounts in the report are net: the overhang grafted onto an end,
less any contig bases trimmed to make room for it. So
Original bp + Total +bp = Final bp for every row, and you can check the report
against the sequences it describes.
After manually extending contigs the revised assembly should be re-polished using available long and short read data to correct indels present in the raw long-reads.
The final telomere-extended assembly should be re-polished using available long and short read data to correct indels (i.e. with NextPolish2 and Pypolca) in the raw long-read extensions.
Optional Quality Control
Additional filters
Users may wish to exclude reads below a minimum mapping quality score to reduce the risk of incorrect alignments.
Similarly, multi-mapping reads will generate secondary alignments. To exclude non-specific aligments you can pre-filtering with samtools view. You can decode sam flags here.
Note: As of version teloclip v0.3.0, filter and extract will exclude secondary alignments by default.
# Use samtools to filter reads below a MAPQ 30
samtools view -h -q 30 input.sam | teloclip filter --ref-idx ref.fa.fai > min_mapq_30.sam
# Exclude secondary alignments by filtering with samtools
# Note: Secondary alignments are filtered by default in teloclip >=v0.3.0, use '--keep-secondary' to keep.
samtools view -h -F 0x100 input.sam | teloclip filter --ref-idx ref.fa.fai > no_secondary.sam
Options
The main teloclip command provides global options and sub-commands for specific operations.
Main Command
Run teloclip --help to view the main command options:
Usage: teloclip [OPTIONS] [COMMAND] [ARGS]...
A tool for the recovery of unassembled telomeres from soft-clipped read
alignments.
Options:
--version Show the version and exit.
--help Show this message and exit.
Commands:
extend Extend contigs using overhang analysis from soft-clipped...
extract Extract overhanging reads for each end of each reference contig.
filter Filter SAM file for clipped alignments containing unassembled...
Filter Sub-command Options
Run teloclip filter --help to view the filter command options:
Usage: teloclip filter [OPTIONS] [SAMFILE]
Filter SAM file for clipped alignments containing unassembled telomeric
repeats.
Options:
--ref-idx PATH Path to fai index for reference fasta. Index
fasta using `samtools faidx FASTA`
[required]
--min-clip INTEGER Require clip to extend past ref contig end
by at least N bases. Default: 1
--max-break INTEGER Tolerate max N unaligned bases before contig
end. Default: 50
--motifs TEXT If set keep only reads containing given
motif/s from comma delimited list of
strings. By default also search for reverse
complement of motifs. i.e. TTAGGG,TTAAGGG
will also match CCCTAA,CCCTTAA
--no-rev If set do NOT search for reverse complement
of specified motifs.
--keep-secondary If set, include secondary alignments in
output. Default: Off (exclude secondary
alignments).
--fuzzy If set, tolerate +/- 1 variation in motif
homopolymer runs i.e. TTAGGG ->
T{1,3}AG{2,4}. Default: Off
-r, --min-repeats INTEGER Minimum number of sequential pattern matches
required for a hit to be reported. Default:
1
--min-anchor INTEGER Minimum number of aligned bases (anchor)
required on the non-clipped portion of the
read. Default: 100
--match-anywhere If set, motif match may occur in unclipped
region of reads.
--log-level [debug|info|warning|error]
Logging level (default: INFO).
--logfile PATH Also write log messages to this file (parent
directories are created).
--help Show this message and exit.
Extract Sub-command Options
Run teloclip extract --help to view the extract command options:
Usage: teloclip extract [OPTIONS] [SAMFILE]
Extract overhanging reads for each end of each reference contig. Reads are
always written to output files.
Options:
--ref-idx PATH Path to fai index for reference fasta. Index
fasta using `samtools faidx FASTA`
[required]
--prefix TEXT Use this prefix for output files. Default:
None.
--extract-dir PATH Write extracted reads to this directory.
Default: cwd.
--min-clip INTEGER Require clip to extend past ref contig end
by at least N bases. Default: 1
--max-break INTEGER Tolerate max N unaligned bases before contig
end. Default: 50
--min-anchor INTEGER Minimum anchored alignment length required
(default: 100).
--min-mapq INTEGER Minimum mapping quality required (default:
0).
--keep-secondary If set, include secondary alignments in
output. Default: Off (exclude secondary
alignments).
--include-stats Include mapping quality, clip length, and
motif counts in FASTA headers.
--count-motifs TEXT Comma-delimited motif sequences to count in
overhang regions (e.g., "TTAGGG,CCCTAA").
--fuzzy-count Use fuzzy motif matching allowing ±1
character variation when counting motifs.
--buffer-size INTEGER Number of sequences to buffer before writing
(default: 1000).
--output-format [fasta|fastq] Output format for extracted sequences
(default: fasta).
--report-stats Write extraction statistics to file in
output directory.
--no-mask-overhangs Do not convert overhang sequences to
lowercase.
--log-level [DEBUG|INFO|WARNING|ERROR]
Logging level (default: INFO).
--logfile PATH Also write log messages to this file (parent
directories are created).
--help Show this message and exit.
Extend sub-command options
Run teloclip extend --help to view the extract command options:
Usage: teloclip extend [OPTIONS] BAM_FILE REFERENCE_FASTA
Extend contigs using overhang analysis from soft-clipped alignments.
Options:
--output-fasta PATH Extended FASTA output file
--stats-report PATH Statistics report output file
--exclude-outliers DEPRECATED and ignored. Contigs with
anomalous overhang coverage are now reported
for review rather than silently dropped;
exclude them with --exclude-contigs if you
agree with the assessment.
--outlier-threshold FLOAT Modified z-score above which a contig end is
reported as having anomalous overhang
coverage (default: 3.5)
--min-overhangs INTEGER Minimum supporting overhangs required
(default: 1)
--max-homopolymer INTEGER Maximum homopolymer run length allowed
(default: 500)
--min-extension INTEGER Minimum novel bases an overhang must
contribute to be used (default: 1)
--min-clip INTEGER Require clip to extend past the contig end
by at least N bases (default: 1)
--max-break INTEGER Maximum gap allowed between alignment and
contig end (default: 50)
--min-anchor INTEGER Minimum anchor length required for alignment
(default: 100)
--dry-run Report extensions without modifying
sequences
--count-motifs TEXT Comma-delimited motif sequences to count in
overhang regions (e.g., "TTAGGG,CCCTAA")
--fuzzy-count Use fuzzy motif matching allowing ±1
character variation when counting motifs
--prefix TEXT Prefix for default output filenames
(default: teloclip_extended)
--screen-terminal-bases INTEGER
Number of terminal bases to screen for
motifs in original contigs (default: 0,
disabled)
--exclude-contigs TEXT Comma-delimited list of contig names to
exclude from extension (e.g.,
"chrM,chrC,scaffold_123")
--exclude-contigs-file PATH Text file containing contig names to exclude
(one per line)
--log-level [debug|info|warning|error]
Logging level (default: INFO).
--logfile PATH Also write log messages to this file (parent
directories are created).
--html-report PATH Write a self-contained HTML report showing
every overhang read aligned against the
contig terminus it supports, plus overhang
depth across the assembly.
--html-max-reads INTEGER Maximum overhang reads rendered per contig
end in the HTML report (default: 25). Reads
contributing the most sequence are shown
first.
--overhang-log PATH Write a TSV describing every accepted
overhang read: contig, end, gap from the
contig terminus, clip length and overhang
length.
--help Show this message and exit.
Citing Teloclip
If you use Teloclip in your work please cite this git repo directly and note the release version you used.
Publications using Teloclip
Teloclip has been used to recover and extend telomeric sequences in a wide variety of taxa, including Algae, Plants, Insects, and Fungi.
-
Deng, Y., Zhou, P., Li, F., Wang, J., Xie, K., Liang, H., Wang, C., Liu, B., Zhu, Z., Zhou, W. and Dun, B., 2024. A complete assembly of the sorghum BTx623 reference genome. Plant Communications, 5(6). 🌾
-
He, W., Hu, D., Guo, M., Nie, B., Zhang, G., Jia, Y., Hou, Z., Shu, S., Shao, Y., Simonsen, H.T. and Twamley, A., 2025. The telomere‐to‐telomere genome of Sanicula chinensis unveils genetic underpinnings of low furanocoumarin diversity and content in one basal lineage of Apiaceae. The Plant Journal, 123(1), p.e70311. 🌱
-
Jaiswal, R.K., Garibo Domingo, T., Grunchec, H., Singh, K., Pirooznia, M., Elhaik, E. and Cohn, M., 2025. Subtelomeric elements provide stability to short telomeres in telomerase-negative cells of the budding yeast Naumovozyma castellii. Current Genetics, 71(1), p.19. 🍄
-
Li, J., Chen, Z., Li, K., Tan, J., Sun, J., Deng, X.W., Park, Y., He, H., Deng, Y. and Zhang, X., 2026. Telomere-to-telomere genome assembly and a mutant library empower functional genomics and genetic improvement in Cucurbita moschata. Plant Communications, 7(5). 🌱
-
Liu, Y., Chen, Y., Ren, Z. et al. Two haplotype-resolved telomere-to-telomere genome assemblies of Xanthoceras sorbifolium. Sci Data 12, 791 (2025). 🌿
-
Liu, Y., Zhao, L., Zhang, J., Ju, Q., Fan, X., Li, Z., Zhang, X., Liang, X., Ge, F. and Chen, J., 2026. Gapless genome assembly and evolutionary analysis of Cnidium monnieri (Apiaceae). Genomics Communications, 3(1). 🌱
-
Loos, A., Doykova, E., Qian, J., Kümmel, F., Ibrahim, H., Kiss, L., Panstruga, R. and Kusch, S., 2025. Saprotrophic Arachnopeziza Species as New Resources to Study the Obligate Biotrophic Lifestyle of Powdery Mildew Fungi. Molecular Ecology Resources, p.e70045. 🍄
-
Oberti, H., Sessa, L., Oliveira‐Rizzo, C., Di Paolo, A., Sanchez‐Vallet, A., Seidl, M.F. and Abreo, E., 2025. Novel genomic features in entomopathogenic fungus Beauveria bassiana ILB308: accessory genomic regions and putative virulence genes involved in the infection process of soybean pest Piezodorus guildinii. Pest Management Science, 81(4), pp.2323-2336. 🍄
-
van Westerhoven, A.C., Mehrabi, R., Talebi, R., Steentjes, M.B., Corcolon, B., Chong, P.A., Kema, G.H. and Seidl, M.F., 2024. A chromosome-level genome assembly of Zasmidium syzygii isolated from banana leaves. G3: Genes, Genomes, Genetics, 14(3), p.jkad262. 🍄
-
Wan, L., Deng, C., Liu, B. et al. Telomere-to-telomere genome assemblies of three silkworm strains with long-term pupal characteristics. Sci Data 12, 501 (2025). 🐛
-
Wang, Z.F., Yu, E.P., Fu, L., Deng, H.G., Zhu, W.G., Xu, F.X. and Cao, H.L., 2025. Chromosome-scale assemblies of three Ormosia species: repetitive sequences distribution and structural rearrangement. GigaScience, 14, p.giaf047. 🌿
-
Xu, Z., Wang, G., Zhu, X. et al. Genome assembly of two allotetraploid cotton germplasms reveals mechanisms of somatic embryogenesis and enables precise genome editing. Nat Genet 57, 2028–2039 (2025). 🌱
-
Yang, H.P., Wenzel, M., Hauser, D.A., Nelson, J.M., Xu, X., Eliáš, M. and Li, F.W., 2021. Monodopsis and Vischeria genomes shed new light on the biology of eustigmatophyte algae. Genome biology and evolution, 13(11), p.evab233. 🦠
Issues
Submit feedback to the Issue Tracker
License
Software provided under GPL-3 license.
Star History

1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file teloclip-0.4.0.tar.gz.
File metadata
- Download URL: teloclip-0.4.0.tar.gz
- Upload date:
- Size: 667.9 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: Hatch/1.17.1 {"ci":true,"cpu":"x86_64","distro":{"id":"noble","libc":{"lib":"glibc","version":"2.39"},"name":"Ubuntu","version":"24.04"},"implementation":{"name":"CPython","version":"3.12.13"},"installer":{"name":"hatch","version":"1.17.1"},"openssl_version":"OpenSSL 3.0.13 30 Jan 2024","python":"3.12.13","system":{"name":"Linux","release":"6.17.0-1020-azure"}} HTTPX2/2.9.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a1ca9dee7d8794dca717400d55e7ddab958417ecd727ce4ffb3086ea83075789
|
|
| MD5 |
5fc756de483727ff4d4fa23dad72ba7a
|
|
| BLAKE2b-256 |
c1423ff1f44487d24cf3f524db50dd611e90df61933bf15e410c502f59edb10d
|
File details
Details for the file teloclip-0.4.0-py3-none-any.whl.
File metadata
- Download URL: teloclip-0.4.0-py3-none-any.whl
- Upload date:
- Size: 109.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: Hatch/1.17.1 {"ci":true,"cpu":"x86_64","distro":{"id":"noble","libc":{"lib":"glibc","version":"2.39"},"name":"Ubuntu","version":"24.04"},"implementation":{"name":"CPython","version":"3.12.13"},"installer":{"name":"hatch","version":"1.17.1"},"openssl_version":"OpenSSL 3.0.13 30 Jan 2024","python":"3.12.13","system":{"name":"Linux","release":"6.17.0-1020-azure"}} HTTPX2/2.9.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ad98d629556488002b87ed6256042ca611d02d8a0c7986db52f630144afc764d
|
|
| MD5 |
83a9463616ea2e138087583475a7cd60
|
|
| BLAKE2b-256 |
746b669b15209eefab83070a32696e1863ff08d6ad1a0cc78bb94badc646f1ff
|









