traitar - The microbial trait analyzer

Project description
Traitar – the microbial trait analyzer
Traitar is a software for characterizing microbial samples from nucleotide or protein sequences. It can accurately phenotype 67 diverse traits.
Table of Contents
Installation
Please see INSTALL.md for installation instructions.
Basic usage
traitar phenotype <in dir> <sample file> from_nucleotides <out_dir>
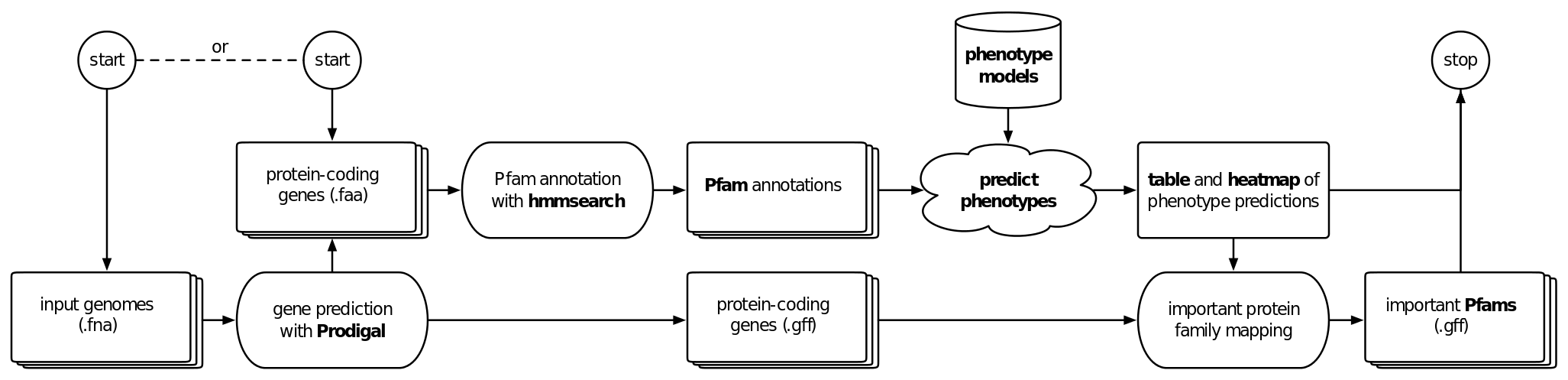
will trigger the standard workflow of Traitar, which is to predict open reading frames with Prodigal, annotate the coding sequences provided as nucleotide FASTAs in the for all samples in with Pfam families using HMMer and finally predict phenotypes from the models for the 67 traits.
The sample file has one column for the sample file names and one for the names as specified by the user. You can also specify a grouping of the samples in the third column, which will be shown in the generated plots. The template looks like following - The header row is mandatory; please also take a look at the sample file for the packaged example data:
traitar phenotype <in dir> <sample file> from_genes <out_dir>
assumes that gene prediction has been conducted already externally. In this case analysis will start with the Pfam annotation. If the output directory already exists, Traitar will offer to recompute or resume the individual analysis steps. This option is only available if the process is run interactively.
Parallel usage
Traitar can benefit from parallel execution. The -c parameter sets the number of processes used e.g. -c 2 for using two processes
traitar phenotype <in dir> <sample file> from_nucleotides out_dir -c 2
This requires installing GNU parallel as noted above.
Inspect phenotype classification models
Traitar can be used to inspect the protein families in each phenotype model:
traitar show 'Glucose fermenter
will show the majority features i.e. the Pfam families that contribute to the assignment of the trait Glucose fermenter with phypat classifier to some genome sequence. Via –predictor the user may specify the classifier (phypat, phypat+PGL).
Run Traitar with packaged sample data
traitar phenotype <traitar_dir>/data/sample_data <traitar_dir>/data/sample_data/samples.txt from_genes <out_dir> -c 2 will trigger phenotyping of Listeria grayi DSM_20601 and Listeria ivanovii WSLC3009. Computation should be done within 5 minutes. You can find out <traitar_dir> by running
``` python python >>> import traitar >>> traitar.path ```
Results
Traitar provides the gene prediction results in <out_dir>/gene_prediction, the Pfam annotation in <out_dir>/pfam_annotation and the phenotype prediction in<out_dir>/phenotype prediction.
Heatmaps
The phenotype prediction is summarized in heatmaps individually for the phyletic pattern classifier in heatmap_phypat.png, for the phylogeny-aware classifier in heatmap_phypat_ggl.png and for both classifiers combined in heatmap_comb.png and provide hierarchical clustering dendrograms for phenotypes and the samples.
Phenotype prediction - Tables and flat files
These heatmaps are based on tab separated text files e.g. predictions_majority-vote_combined.txt. A negative prediction is encoded as 0, a prediction made only by the pure phyletic classifier as 1, one made by the phylogeny-aware classifier by 2 and a prediction supported by both algorithms as 3. predictions_flat_majority-votes_combined.txt provides a flat version of this table with one prediction per row. The expert user might also want to access the individual results for each algorithm in the respective sub folders phypat and phypat+PGL.
Phenotype-relevant protein families and feature tracks
Traitar will link the protein families and predicted phenotypes. The results can be found in phypat/feat_gffs and `phypat+PGL/feat_gffs. If the user picked the ‘from nucleotides’ option, Traitar will also generate GFF files that link the genes called by Prodidgal with the important protein families. The phenotype-specific protein family annotations tracks can be visualized via GFF files in a genome browser of choice.
Feature tracks with from_genes option (experimental feature)
If the from_genes option is set, the user may specify gene GFF files via an additional column called gene_gff in the sample file. As gene ids are not consistent across gene GFFs from different sources e.g. img, RefSeq or Prodigal the user needs to specify the origin of the gene gff file via the -g / –gene_gff_type parameter. Still there is no guarantee that this works currently. Using samples_gene_gff.txt as the sample file in the above example will generate phenotype-specific Pfam tracks for the two genomes.
traitar phenotype . samples_gene_gff.txt from_genes traitar_out -g refseq
Docker
There is a Docker container available for Traitar. Pull by
docker pull aweimann/traitar
To run traitar for the sample data execute
docker run -v <traitar_dir>/data/sample_data:/mnt 1445e6c01992 bash -c 'traitar phenotype /mnt/ /mnt/samples.txt from_nucleotides /mnt/traitar_out',
which will take ~30 minutes. Note there is a problem with parallel usage so -c option is not guaranteed to work. The output will be owned by root. So currently you still need root access to your machine to inspect the traitar_out folder.
Citing Traitar
If you use Traitar in your research, please cite our preprint:


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file traitar-1.1.2.tar.gz.
File metadata
- Download URL: traitar-1.1.2.tar.gz
- Upload date:
- Size: 8.4 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
73886d01c7431cb9bd4345192920fad0a430aeae48ba5b4b844f843f809e5387
|
|
| MD5 |
cfd558771c0177ca4aa365906e30f5c3
|
|
| BLAKE2b-256 |
8a10b90d7e3498fbfd314ca1edccf88eb47e3ef5ad8c9acb46ed16465557dd45
|








{kind=link}
{kind=link}