Python implementation of the spectrophore descriptor

Project description
Spectrophores


This module contains python code to calculate spectrophores from molecules. It is uses RDKit to process molecules, and numba to improve the performance.
The technology and its applications have been described in Journal of Cheminformatics (2018) 10, 9. The paper is also included in this distribution.
The spectrophore code can be used in two ways:
- As a standalone program to convert the molecules in a sd-file into their corresponding
spectrophores - As a
pythonmodule to import in your ownpythoncode.
In the following sections, both usages will be documented.
This repository also includes spectrophore_notebook.ipynb, a portable Jupyter Notebook which could be useful for quick and interactive data analysis with spectrophores.
Installation
Installation using pip (recommended)
-
Create and activate a new Anaconda environment
conda create --name spectrophore python=3conda activate spectrophore -
Install the repo via pip. The RDKit, Numba and other dependencies should be installed automatically.
pip install uamc-spectrophore
Installation from Source
-
Clone the repository and navigate to it
git clone https://github.com/silicos-it/spectrophore.gitcd spectrophore -
Create and activate a new Anaconda environment
conda create --name spectrophore python=3conda activate spectrophore -
Install the repo by using pip. The RDKit, Numba and other dependencies should be installed automatically.
pip install .For Contributors
To install with additional development dependencies (
pytest,ruff, etc.), use:pip install .[develop]
Usage
1. Usage as a standalone program
After installation, one should be able to use the spectrophore package as a standalone program to calculate spectrophores from a sd-file with molecules.
> python -m spectrophore -h
or even more convenient:
> spectrophore -h
This will provide you with all details on how to calculate spectrophores from a sd-file, as well as all the configurable settings:
usage: spectrophore [-h] [-n {none,mean,all,std}] [-m {classic,full}] [-s {none,unique,mirror,all}] [-a {1,2,3,4,5,6,9,10,12,15,18,20,30,36,45,60,90,180}] [-r RESOLUTION] [-p MAX_WORKERS]
[--silent] -i INFILE -o OUTFILE
Convert a SDF-file containing 3D structures to a csv with the corresponding spectrophores fingerprints
options:
-h, --help show this help message and exit
-n {none,mean,all,std}, --norm {none,mean,all,std}
Normalization setting (default: all)
-m {classic,full}, --mode {classic,full}
Which type of spectrophore to calculate (default: classic)
-s {none,unique,mirror,all}, --stereo {none,unique,mirror,all}
Stereo setting (default: none)
-a {1,2,3,4,5,6,9,10,12,15,18,20,30,36,45,60,90,180}, --accuracy {1,2,3,4,5,6,9,10,12,15,18,20,30,36,45,60,90,180}
Accuracy setting (default: 20)
-r RESOLUTION, --resolution RESOLUTION
Resolution setting (>0) (default: 3)
-p MAX_WORKERS, --np MAX_WORKERS
Number of processors to use; -1 is all processors (default: -1)
--silent Don't show a progressbar (default: False)
required arguments:
-i INFILE, --in INFILE
Input sdf file (default: None)
-o OUTFILE, --out OUTFILE
Output spectrophore file (default: None)
2. Usage as a python module
2.1. Introduction
Once you have installed the uamc-spectrophore package, you are ready to calculate spectrophores. In its most simple form, spectrophores can be computed as follows:
>>> from spectrophore import spectrophore
>>> from rdkit import Chem
>>> from rdkit.Chem import AllChem
>>> mol = Chem.MolFromSmiles("c1ncncc1")
>>> mol = Chem.AddHs(mol)
>>> AllChem.EmbedMolecule(mol, randomSeed=1)
0
>>> calculator = spectrophore.SpectrophoreCalculator(normalization='none')
Probes initialised: 48 number of probes in total
12 probes are used due to the imposed stereo flag
>>> calculator.calculate(mol)
array([ 1.4826062, 2.0534244, 1.6182035, 3.1277552, 2.5039089,
5.157424 , 2.3022916, 1.6652936, 3.4908977, 4.0566797,
5.020808 , 3.0757458, 0.6589167, 0.7786141, 4.324721 ,
4.3273506, 2.928256 , 4.132863 , 1.9475778, 3.5233636,
4.6044445, 4.1554613, 4.5122004, 5.2647495, 46.877754 ,
71.05832 , 96.21371 , 94.29615 , 51.998947 , 63.523525 ,
24.393661 , 80.728264 , 73.68167 , 65.27555 , 56.712753 ,
102.68948 , 0.7165117, 1.157669 , 2.1900623, 2.189616 ,
1.0546725, 1.2338142, 0.5119512, 1.7162334, 1.6656369,
1.2711622, 1.2308022, 2.2867224], dtype=float32)
In the example shown, the first three lines import the required modules: module spectrophore for the calculation of spectrophores, module Chem to generate a RDKit molecule from a smiles string, and module AllChem to generate a 3D-conformation from the molecule. Next, a molecule is created from a smiles string (line 4), and a conformation is then generated at the 6th line after adding hydrogen atoms on the 5th line. Finally, on lines 7 and 8, a SpectrophoreCalculator object is generated and this object is then used to calculate a spectrophore descriptor (line 8), which consists in its default form of 4 * 12 numbers.
Note: a few words on the shape of a
spectrophore:Each
spectrophoreconsists of a set of floating point numbers, and this set is always a multiple of 4 (the number of properties). The actual number count depends on how stereochemistry is treated in the calculation of spectrophores; this is controlled by thestereo()method:
stereo("none"): the total number of numbers in aspectrophoreis 48 (4 * 12; default),stereo("unique"): the total number of numbers in aspectrophoreis 72 (4 * 18),stereo("mirror"): the total number of numbers in aspectrophoreis 72 (4 * 18),stereo("all"): the total number of numbers in aspectrophoreis 144 (4 * 36).
Whatever the actual number of
spectrophorepoints, these are always calculated according the same atomic properties. For example, consider aspectrophoreof 4n points, then these points represent the following:
- Points 1 to n: representing the interaction energies between the atomic partial charges and each of the n boxes;
- Points n+1 to 2n: representing the interaction energies between the atomic lipophilicities and each of the n boxes;
- Points 2n+1 to 3n: representing the interaction energies between the atomic shape deviations and each of the n boxes;
- Points 3n+1 to 4n: representing the interaction energies between the atomic electrophilicities and each of the n boxes.
Please have a look at the original publication form more information about the way these interaction energies are calculated, and what the
stereo()method actually means.
If a molecule contains more than one 3D-conformation, then one may specify which conformation should be used for the calculation of spectrophores. As an example, consider the following code:
>>> calculator = spectrophore.SpectrophoreCalculator(normalization='none')
Probes initialised: 48 number of probes in total
12 probes are used due to the imposed stereo flag
>>> aspirin = Chem.MolFromSmiles("CC(Oc1ccccc1C(O)=O)=O")
>>> aspirin = Chem.AddHs(aspirin)
>>> cids = AllChem.EmbedMultipleConfs(aspirin, numConfs=3, randomSeed=1)
>>> print(len(cids))
3
>>> for i in range(len(cids)): calculator.calculate(aspirin, i)
...
array([ 3.9325106, 5.057322 , 3.4856937, 5.479296 , 5.4948115,
6.778553 , 3.3464622, 4.14602 , 5.3836374, 7.561534 ,
6.8232827, 5.989737 , 7.6784873, 8.210138 , 7.167798 ,
10.159893 , 21.949173 , 21.82916 , 14.773877 , 17.42354 ,
19.190207 , 23.495762 , 27.787817 , 16.30314 , 216.26787 ,
333.40674 , 342.49576 , 360.4995 , 314.15094 , 339.17233 ,
154.0079 , 332.78748 , 327.1066 , 362.64258 , 318.9287 ,
409.1051 , 1.9632099, 3.081648 , 3.7438314, 4.0490594,
3.3057344, 3.7012224, 1.5342569, 3.5204902, 3.414244 ,
3.5912938, 3.358138 , 4.4090977], dtype=float32)
array([ 3.3760934, 6.2265506, 4.525822 , 4.819302 , 5.8221617,
5.351173 , 4.5672374, 5.7664223, 6.5604424, 8.637403 ,
8.085242 , 5.02033 , 6.9545755, 7.845039 , 7.3663807,
10.209895 , 22.197407 , 20.808294 , 14.168869 , 17.2765 ,
18.936844 , 23.069283 , 26.431952 , 16.029125 , 194.90318 ,
353.951 , 286.0087 , 303.20142 , 244.75073 , 268.89917 ,
169.84006 , 299.00415 , 282.5781 , 321.46698 , 344.0466 ,
359.3506 , 1.7995539, 3.2626312, 3.2555926, 3.4746647,
2.682269 , 2.511837 , 1.6443442, 3.2317195, 2.7628746,
3.0616467, 3.5081995, 3.877851 ], dtype=float32)
array([ 3.6380727, 5.0543313, 3.4793944, 5.1971545, 5.2814097,
6.6500306, 3.5975058, 3.8349445, 5.3234606, 7.27565 ,
6.456969 , 5.91822 , 7.750357 , 8.797789 , 7.37978 ,
10.291023 , 20.842691 , 21.349953 , 14.5347595, 17.440334 ,
19.158358 , 23.11196 , 27.614624 , 15.990323 , 204.78897 ,
325.7338 , 321.78525 , 338.5145 , 267.19482 , 374.50073 ,
142.66623 , 303.51685 , 296.4746 , 343.62027 , 324.18774 ,
394.01352 , 1.8762052, 3.0206196, 3.4741364, 3.7698758,
2.7347808, 3.719328 , 1.4108949, 3.237199 , 3.2545724,
3.386793 , 3.4655266, 4.238542 ], dtype=float32)
One can easily visualise spectrophores by plotting the actual values using matplotlib. For example, consider the following snippet:
>>> import matplotlib.pyplot as plt
>>> mol = Chem.MolFromSmiles("CC(CCC1=CC=CC=C1Cl)N1CCOCC1")
>>> mol = Chem.AddHs(mol)
>>> cids = AllChem.EmbedMultipleConfs(mol, numConfs = 10, randomSeed = 1)
>>> spectrophores = []
>>> for cid in cids: spectrophores.append(calculator.calculate(mol, cid))
...
>>> for i in range(len(spectrophores)): plt.plot(range(1,49), spectrophores[i], label='Conf %d' % (i+1))
...
[<matplotlib.lines.Line2D object at 0x7f989dc3f4f0>]
[<matplotlib.lines.Line2D object at 0x7f989dc3f850>]
[<matplotlib.lines.Line2D object at 0x7f989dc3fbb0>]
[<matplotlib.lines.Line2D object at 0x7f989dc3ff10>]
[<matplotlib.lines.Line2D object at 0x7f989dc512b0>]
[<matplotlib.lines.Line2D object at 0x7f989dc51610>]
[<matplotlib.lines.Line2D object at 0x7f989dc51970>]
[<matplotlib.lines.Line2D object at 0x7f989dc51cd0>]
[<matplotlib.lines.Line2D object at 0x7f989dc5e070>]
[<matplotlib.lines.Line2D object at 0x7f989dc5e3d0>]
>>> plt.legend(loc='upper left')
<matplotlib.legend.Legend object at 0x7f9899470e80>
>>> plt.grid()
>>> plt.savefig("spectrophore/images/exampleplot1.png")
which generates the following plot:
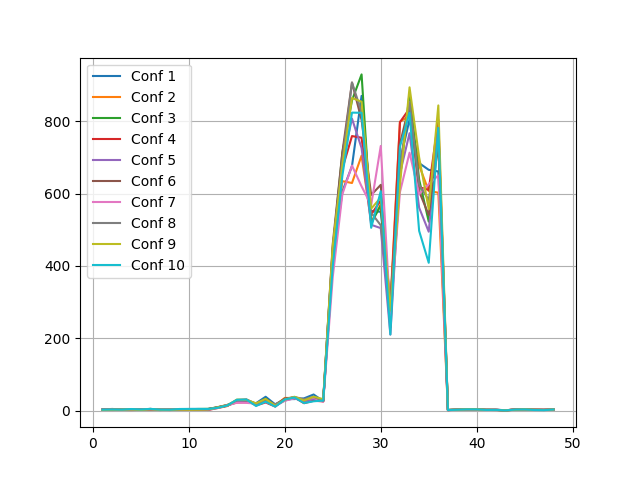
Similarly, one can easily compare the spectrophores from two different molecules, and quantify the difference:
>>> plt.close()
>>> spectrophores = []
>>> mols = [Chem.MolFromSmiles("ClC(Br)(I)F"), Chem.MolFromSmiles("CC(CCC1=CC=CC=C1Cl)N1CCOCC1")]
>>> for i in range(2):
... mols[i] = Chem.AddHs(mols[i])
... AllChem.EmbedMolecule(mols[i], randomSeed=1)
... spectrophores.append(calculator.calculate(mols[i]))
...
0
0
>>> for i in range(2): plt.plot(range(1,49), spectrophores[i], label='Molecule %d' % (i+1))
...
[<matplotlib.lines.Line2D object at 0x7faf420df520>]
[<matplotlib.lines.Line2D object at 0x7faf420df4c0>]
>>> plt.grid()
>>> plt.savefig("spectrophore/images/exampleplot2.png")
>>> from scipy.spatial import distance
>>> distance.euclidean(spectrophores[0],spectrophores[1])
2129.031982421875
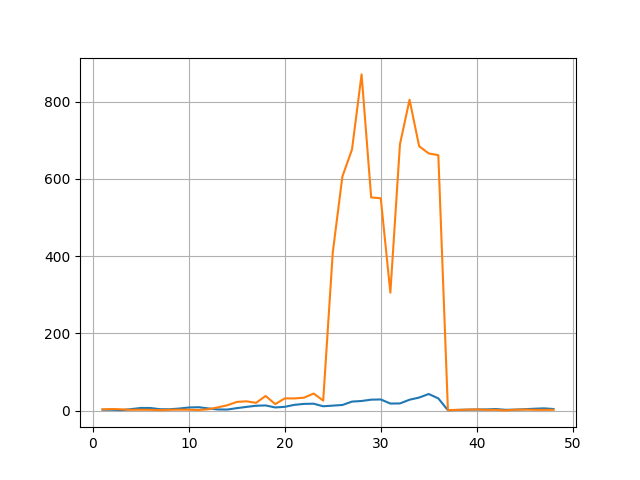
From the last example, it is clear that the actual spectrophore values may differ a lot depending on the type of molecule. Also, the absolute values are depending on the property type, with some properties leading to large values (e.g. shape deviation) and others very small. For this reason, a number of normalization methods are provided as shown below.
2.2. Module methods
resolution
The resolution() method controls the smallest distance between the molecule and the surrounding box. By default this value is set to 3.0 A. The resolution() can be specified at the moment of class creation, or later on using the resolution() method:
>>> mol = Chem.MolFromSmiles("ClC(Br)(I)F")
>>> AllChem.EmbedMolecule(mol, randomSeed=1)
0
>>> calculator = spectrophore.SpectrophoreCalculator(normalization='none') # Default of 3.0
Probes initialised: 48 number of probes in total
12 probes are used due to the imposed stereo flag
>>> print(calculator.calculate(mol)[0])
2.9870005
>>> calculator = spectrophore.SpectrophoreCalculator(normalization='none', resolution = 5.0)
Probes initialised: 48 number of probes in total
12 probes are used due to the imposed stereo flag
>>> print(calculator.calculate(mol)[0])
1.3418818
>>> calculator.resolution(10.0)
>>> print(calculator.calculate(mol)[0])
0.33471847
The larger the resolution value (e.g. 10.0 versus 3.0 A), the smaller the interaction energies and corresponding spectrophore values.
Calling the resolution() method without an argument returns the current resolution value:
>>> calculator.resolution()
10.0
accuracy
The accuracy() method controls the angular stepsize by which the molecule is rotated within the cages. By default this value is set to 20°. This parameter can be modified either at class creation, or using the accuracy() method later on. The accuracy should be an integer fraction of 180, hence 180 modulus accuracy should be equal to 0. The smaller the accuracy value (meaning smaller angular stepsizes), the longer the computation time:
>>> calculator = spectrophore.SpectrophoreCalculator(accuracy = 20.0, normalization = 'none') # Default
Probes initialised: 48 number of probes in total
12 probes are used due to the imposed stereo flag
>>> print(calculator.calculate(mol)[0])
2.9870005
>>> calculator = spectrophore.SpectrophoreCalculator(accuracy = 2.0, normalization = 'none')
Probes initialised: 48 number of probes in total
Only using 12 probes
>>> print(calculator.calculate(mol)[0]) # Takes some time
3.0315506
>>> 100.0 * (3.0315506 - 2.9870005) / 3.0315506
1.469548289908139
Calling the accuracy() method without an argument returns the current accuracy value:
>>> calculator.accuracy()
np.int64(2)
normalization
With the normalization() method, one can specify the type of spectrophore normalization. There are four possibilities:
normalization("none"): no normalization is applied and thespectrophorevalues are the raw calculated interaction energies (multiplied by -100),normalization("mean"): for each property, the average value is calculated and each of the individualspectrophoreproperty value are reduced by these mean values. This centers the calculated values around 0,normalization("std"): for each property, the standard deviation is calculated and each of the individualspectrophoreproperty value is divided by these standard deviations, making the standard deviation 1normalization("all"): each spectrophore value is normalized by mean and standard deviation. This is the default option.
The default value is "all".
>>> calculator.accuracy(20)
>>> calculator.normalization("none")
>>> spec = calculator.calculate(mol)
>>> print(spec[:12])
[2.9870005 2.7023234 1.8029699 4.468908 7.375546 7.252276 4.1123323
4.0559926 5.908458 8.5649605 9.328256 6.2296877]
>>> sum(spec[:12])
np.float32(64.78871)
>>> calculator.normalization("mean")
>>> spec = calculator.calculate(mol)
>>> print(spec[:12])
[-2.4120588 -2.6967359 -3.5960894 -0.93015146 1.9764867 1.8532166
-1.286727 -1.3430667 0.50939894 3.1659012 3.9291964 0.8306284 ]
>>> sum(spec[:12])
np.float32(-4.7683716e-07)
>>> calculator.normalization("std")
>>> spec = calculator.calculate(mol)
>>> print(spec[:12])
[1.2924119 1.1692382 0.78010696 1.933602 3.1912427 3.1379066
1.7793193 1.7549422 2.556465 3.7058773 4.036139 2.6954541 ]
>>> sum(spec[:12])
np.float32(28.032705)
>>> calculator.normalization("all")
>>> spec = calculator.calculate(mol)
>>> print(spec[:12])
[-1.0436468 -1.1668205 -1.5559518 -0.40245688 0.855184 0.80184764
-0.55673957 -0.58111656 0.22040614 1.3698184 1.7000802 0.35939535]
>>> sum(spec[:12])
np.float32(-6.2584877e-07)
Using a normalization over 'all' makes it more easier to compare spectrophores between molecules:
>>> plt.close()
>>> mols = [Chem.MolFromSmiles("ClC(Br)(I)F"), Chem.MolFromSmiles("CC(CCC1=CC=CC=C1Cl)N1CCOCC1")]
>>> spectrophores = []
>>> for i in range(2):
... mols[i] = Chem.AddHs(mols[i])
... AllChem.EmbedMolecule(mols[i], randomSeed = 1)
... spectrophores.append(calculator.calculate(mols[i]))
...
0
0
>>> for i in range(2): plt.plot(range(1,49), spectrophores[i], label='Molecule %d' % (i+1))
...
[<matplotlib.lines.Line2D object at 0x744aeb797770>]
[<matplotlib.lines.Line2D object at 0x744aeb797aa0>]
>>> plt.legend()
<matplotlib.legend.Legend object at 0x744af8519640>
>>> plt.grid()
>>> plt.savefig("spectrophore/images/exampleplot3.png")
>>> from scipy.spatial import distance
>>> distance.euclidean(spectrophores[0],spectrophores[1])
8.419559478759766
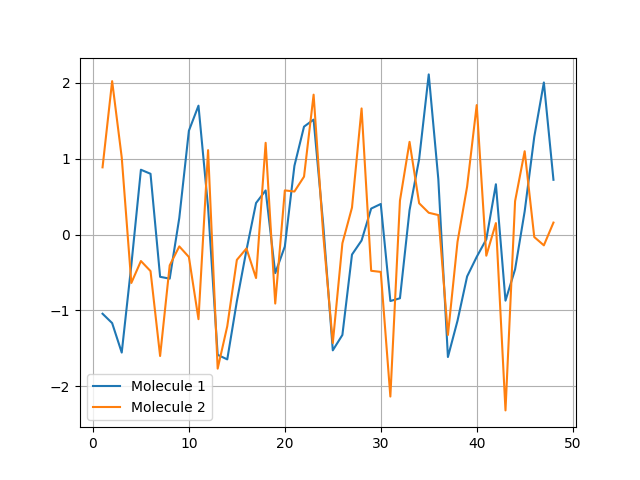
The same holds true when comparing spectrophores from different conformations:
>>> plt.close()
>>> spectrophores = []
>>> mol = Chem.MolFromSmiles("CC(CCC1=CC=CC=C1Cl)N1CCOCC1")
>>> mol = Chem.AddHs(mol)
>>> cids = AllChem.EmbedMultipleConfs(mol, numConfs = 10, randomSeed = 1)
>>> calculator.normalization("all")
>>> for cid in cids: spectrophores.append(calculator.calculate(mol, cid))
...
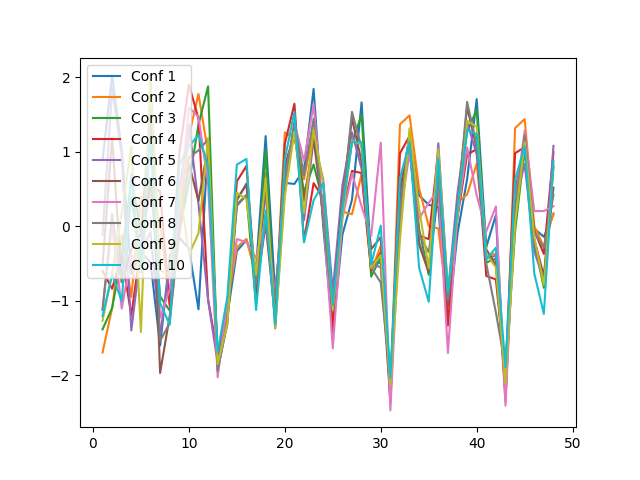
>>> for i in range(len(spectrophores)): plt.plot(range(1,49), spectrophores[i], label='Conf %d' % (i+1))
...
[<matplotlib.lines.Line2D object at 0x744aeb7ee000>]
[<matplotlib.lines.Line2D object at 0x744aeb7c6780>]
[<matplotlib.lines.Line2D object at 0x744aeb7c43e0>]
[<matplotlib.lines.Line2D object at 0x744aeb7c5eb0>]
[<matplotlib.lines.Line2D object at 0x744aeb7c51f0>]
[<matplotlib.lines.Line2D object at 0x744aeb7c5340>]
[<matplotlib.lines.Line2D object at 0x744aeb7c5a30>]
[<matplotlib.lines.Line2D object at 0x744aeb7c6180>]
[<matplotlib.lines.Line2D object at 0x744aeb7c5e20>]
[<matplotlib.lines.Line2D object at 0x744aeb7c70e0>]
>>> plt.legend(loc='upper left')
<matplotlib.legend.Legend object at 0x744aeb8291c0>
>>> plt.savefig("spectrophore/images/exampleplot4.png")
>>> from scipy.spatial import distance
>>> distance.euclidean(spectrophores[0],spectrophores[1])
6.242583751678467
>>> distance.euclidean(spectrophores[0],spectrophores[2])
6.020205497741699
>>> distance.euclidean(spectrophores[1],spectrophores[2])
4.005795001983643
stereo
The stereo() method specifies the kind of cages to be used. The reason for this is that some of the cages that are used to calculate spectrophores have a stereospecific distribution of the interaction points:
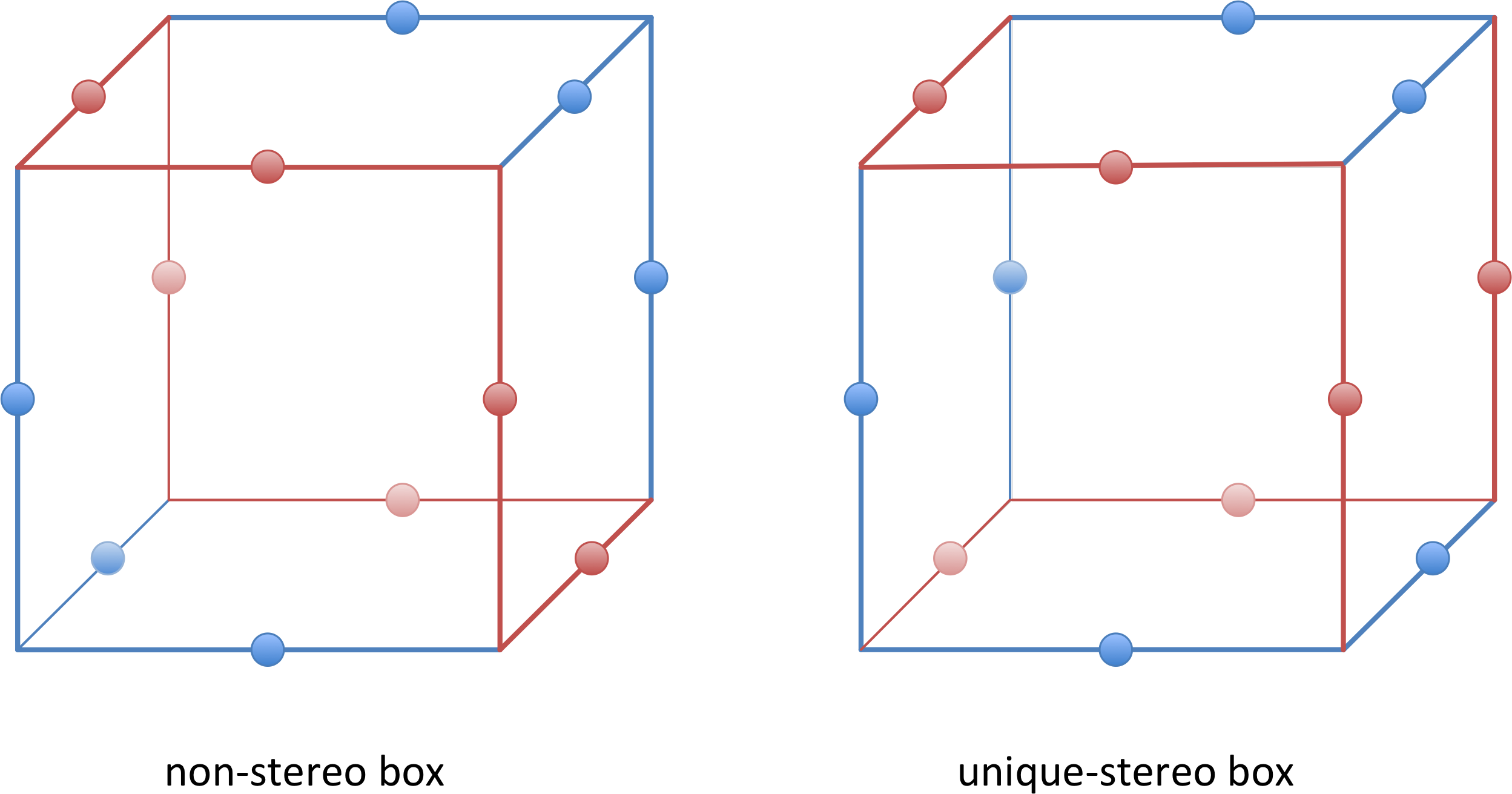
There are four possibilities:
stereo("none"): no stereospecificity (default).Spectrophoresare generated using cages that are not stereospecific. For most applications, thesespectrophoreswill sufficestereo("unique"): unique stereospecificity.Spectrophoresare generated using unique stereospecific cagesstereo("mirror"): mirror stereospecificity. Mirror stereospecificspectrophoresarespectrophoresresulting from the mirror enantiomeric form of the input moleculesstereo("all"): all cages are used. This results in the longestspectrophoresand should only in specific cases be used
The differences between the corresponding data points of unique and mirror stereospecific spectrophores are very small and require very long calculation times to obtain a sufficiently high quality level. This increased quality level is triggered by the accuracy setting and will result in calculation times being increased by at least a factor 100. As a consequence, it is recommended to apply this increased accuracy only in combination with a limited number of molecules, and when the small differences between the stereospecific spectrophores are really critical. However, for the vast majority of virtual screening applications, this increased accuracy is not required as long as it is not the intention to draw conclusions about differences in the underlying molecular stereoselectivity. Non-stereospecific spectrophores will therefore suffice for most applications.
mode
The mode() method currently has two supported options:
mode("classic"): This is the default option, and computes spectrophores as described in the original manuscript.mode("full"): This option keeps all the data from every frame, which could potentially be useful in down-stream applications. The output is a 2D NumPy array, where:- The first dimension corresponds to the number of frames, which is determined by the accuracy option.
- The second dimension represents the length of the spectrophore, which is influenced by the stereo option.
Computing the column-wise minimum of a full spectrophore yields the corresponding classic spectrophore
3. Interpreting spectrophores
A spectrophore is a vector of real number and has a certain length. The length depends on the used stereo method and the number of properties. The standard setting uses a set of non-stereospecific probes in combination with four properties:
- property 1: atomic partial charges
- property 2: atomic lipophilicities
- property 3: atomic shape deviations
- property 4: atomic electrophilicties
The combination of four properties and the set of non-stereospecific probes leads to a spectrophore vector length of 48. The use of other probes leads to other vector lengths, as summarised in this table:
| Stereospecificity | Number of probes | Number of properties | Length |
|---|---|---|---|
| none | 12 | 4 | 48 |
| unique | 18 | 4 | 72 |
| mirror | 18 | 4 | 72 |
| all | 36 | 4 | 144 |
The general layout of a spectrophore, irrespective of its length, is always:
| Property 1 | Property 2 | Property 3 | Property 4 |
|---|---|---|---|
| probe 1..probe n | probe 1..probe n | probe 1..probe n | probe 1..probe n |
meaning that the first n values (with n being the number of probes) are calculated using property 1 (partial charges), then another n values (n+1 up to 2n) calculated using property 2 (lipophilicities), and so forth.
4. Release updates
-
1.3.0:
- Added the mode() method to the SpectrophoreCalculator class, with 'classic' and 'full' as options
- Added the calculate_string() method to the SpectrophoreCalculator class
- Added CI using GitHub Actions
- Updated setup.py for easier install using pip
- Moved the main function to a seperate main.py, making the standalone functionality more easily accessible using the spectrophore alias
- Moved the Jupyter notebook to the main repo and updated it to the new code
- Updated readme
- General code cleanup
-
1.2.0:
- Switched to numpy.float32 type to achieve major speedup
-
1.1.0:
- Updated and optimised the NumPy code
- Bug fixes
- Introduced Numba
- Made the 'all' normalization method the default one
- Added a test suite
-
1.0.1: First official release on PyPi
5. Reference and citation
If you use the spectrophore technology in your own research work, please cite as follows:
Gladysz, R.; Mendes Dos Santos, F.; Langenaeker, W.; Thijs, G.; Augustyns, K.; De Winter, H. (2018) 'Spectrophores as one-dimensional descriptors calculated from three-dimensional atomic properties: applications ranging from scaffold hopping to multi-target virtual screening', J. Cheminformatics 10, 9.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file uamc_spectrophore-1.3.0.tar.gz.
File metadata
- Download URL: uamc_spectrophore-1.3.0.tar.gz
- Upload date:
- Size: 40.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.12.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
571f50d854bc7ce2d8abfe4e5c19784f479ff3b99e38cf0019db382cc7766414
|
|
| MD5 |
2f28e041309d3d16bad952ee8ef9740b
|
|
| BLAKE2b-256 |
7363c87d71194c934214ec0c14de3725082ea9d0b3fad38a2583750fa746ab88
|
File details
Details for the file uamc_spectrophore-1.3.0-py3-none-any.whl.
File metadata
- Download URL: uamc_spectrophore-1.3.0-py3-none-any.whl
- Upload date:
- Size: 30.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.12.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5145b57de7880bc4c3300127270f098bf28c3d74b9a774c036a7d42ceaee8098
|
|
| MD5 |
7639dc9088916946d6890503acc78a38
|
|
| BLAKE2b-256 |
9e63464daa14f7bdf4ac7a89af91f328e423906c5b7a67a0f94df7f221c1e39b
|







