This package has functions for the conversion of amino acid sequences to physicochemical vectors and the subsequent analysis of those vector sequences.

Project description
PCDTW is a package that implements the conversion of amino acid sequences to physicochemical vectors and subsequently allows for alignment of the sequences based on those vectors, development of consensus vectors that can be used to search databases for similar physicochemical profiles, development of the DTW distance between two physicochemical vectors and a few other functions. The basis for this package can be found in three publications and should be consulted for further background [1–3].
To install PCDTW (Two Options): -Use ‘pip install PCDTW’ in a powershell prompt -Use ‘!pip install PCDTW’ in a jupyter notebook
To use PCDTW: Use ‘import PCDTW’
Citations
1)Dixson, J.D.; Vumma, L.; Azad, R.K. An Analysis of Combined Molecular Weight and Hydrophobicity Similarity between the Amino Acid Sequences of Spike Protein Receptor Binding Domains of Betacoronaviruses and Functionally Similar Sequences from Other Virus Families. Microorganisms 2024, 12.
2)Dixson, J.D.; Azad, R.K. Physicochemical Evaluation of Remote Homology in the Twilight Zone. Proteins Struct. Funct. Bioinforma. 2024, n/a, doi:https://doi.org/10.1002/prot.26742.
3)Dixson, J.D.; Azad, R.K. A Novel Predictor of ACE2-Binding Ability among Betacoronaviruses. Evol. Med. Public Heal. 2021, 9, 360–373, doi:10.1093/EMPH/EOAB032.
Usage:
-
To convert an amino acid sequence to vector form using two physicochemical properties:
PCDTW.PCDTWConvert(x, PCProp1='Mass', PCProp2='HydroPho', normalize=True, NormType='MinMax')
PCProp1/PCProp2 options:
- 'HydroPho'
- 'HydroPhIl'
- 'Hbond'
- 'SideVol'
- 'Polarity'
- 'Polarizability'
- 'SASA'
- 'NCI'
- 'Mass'
- 'None'
Normalization: If normalize is set to True then the individual physicochemical scalar values for each amino acid are normalized before converting the amino acid sequence to vector form. Normalization can be set to 'MinMax' or 'AbsMax'.
-
To align two amino acid sequences using DTW and two physicochemical properties:
PCDTW.PCDTWPWAlign(inputseq1str, inputseq2str, PCProp1='Mass', PCProp2='HydroPho', Penalty=0, Window=3, GAP="Gap")
Window= size of Sakoe-Chiba bandPenalty= somewhat equivalent to mismatch penalty in standard dynamic programming based alignmentGAP= can be 'Gap' or 'Lower' and determines how gaps are presented
Returns a dictionary containing the following values:
- 'Seq1AlignedString'
- 'Seq2AlignedString'
- 'FullAlignment'
- 'Identity'
- 'ConsensusVector'
Example to get the full alignment and identity:
seq1 = "MSDSNQGNNQQNYQQYSQNGNQQQGNNRYQG" seq2 = "MMNNNGNQVSNLSNALRQVNIGNRNSNTTT" PairwiseAlignment=PCDTW.PCDTWPWAlign(seq1, seq2, PCProp1='Mass', PCProp2='HydroPho') print(PairwiseAlignment['FullAlignment']) print(PairwiseAlignment['Identity'])
-
To get the PCDTW distance between two sequences normalized to the number of amino acids in the alignment:
Dist=PCDTW.PCDTWDist(Seq1, Seq2, PCProp1='Mass', PCProp2='HydroPho') print(Dist)
Example to get the distance:
seq1 = "MSDSNQGNNQQNYQQYSQNGNQQQGNNRYQG" seq2 = "MMNNNGNQVSNLSNALRQVNIGNRNSNTTT" print(PCDTWDist(seq1, seq2))
-
To get synthetically evolved homologs for an input sequence:
SynHomologs=PCDTW.PCEvolve(Seq='GALM', PCProp1='Mass', PCProp2='HydroPho', BaseName='ProtX') print(SynHomologs)
PCProp1/PCProp2 options:
- 'HydroPho'
- 'HydroPhIl'
- 'Hbond'
- 'SideVol'
- 'Polarity'
- 'Polarizability'
- 'SASA'
- 'NCI'
- 'Mass'
- 'None'
-
To get a newick format tree using PCDTW that represents the physicochemical similarity of protein sequences:
Newick=PCDTW.PCDTWTree(FastaFile='Your_File_Location.fasta',PCProp1='Mass', PCProp2='HydroPho') print(Newick)
PCProp1/PCProp2 options:
- 'HydroPho'
- 'HydroPhIl'
- 'Hbond'
- 'SideVol'
- 'Polarity'
- 'Polarizability'
- 'SASA'
- 'NCI'
- 'Mass'
- 'None'
This function is derived from the original algorithm used in Dixson and Azad, 2021. Unlike the original algorithm the two physicochemical properties used can be set to any two from the nine included above or 'None' if the user prefers to use only one physicochemical property. If the PCProps are not specified by the user then they default to mass and hydrophobicity. The hydrophobicity values used in this package vary slightly from those used in the original algorithm.
-
To perform multiple sequence alignment:
FASTALoc=Fasta file location as a string MSAAlignment=PCDTW.PCDTWMSAlign(FASTALoc, PCProp1='Mass', PCProp2='HydroPho', n_jobs=-1) print(MSAAlignment[0])#Gives the output as a list of aligned sequences print(MSAAlignment[1])#Gives the output in Fasta format
PCProp1/PCProp2 options:
- 'HydroPho'
- 'HydroPhIl'
- 'Hbond'
- 'SideVol'
- 'Polarity'
- 'Polarizability'
- 'SASA'
- 'NCI'
- 'Mass'
- 'None'
n_jobs options:
- -1 use all available cores
- Integer 1 or greater specifies the number of cores to use
Physicochemical alignments are similar to standard alignments with a few main differences. They are physicochemical equivalency alignments. In other words, they will only directly correspond to the true evolutionary path when the physicochemical properties selected for at each residue position are known. However, some of the more fundamental physicochemical properties have proven to be good general indicators; most notably molecular weight and hydrophobicity.
7)To convert and amino acid sequence to a 45 dimensional vector representing 36 combinations of nine physicochemical properties and the 9 individual properties. The vectors are in the following order: ('HydroPho', 'HydroPhIl'), ('HydroPho', 'Hbond'), ('HydroPho', 'SideVol'), ('HydroPho', 'Polarity'), ('HydroPho', 'Polarizability'), ('HydroPho', 'SASA'), ('HydroPho', 'NCI'), ('HydroPho', 'Mass'), ('HydroPhIl', 'Hbond'), ('HydroPhIl', 'SideVol'), ('HydroPhIl', 'Polarity'), ('HydroPhIl', 'Polarizability'), ('HydroPhIl', 'SASA'), ('HydroPhIl', 'NCI'), ('HydroPhIl', 'Mass'), ('Hbond', 'SideVol'), ('Hbond', 'Polarity'), ('Hbond', 'Polarizability'), ('Hbond', 'SASA'), ('Hbond', 'NCI'), ('Hbond', 'Mass'), ('SideVol', 'Polarity'), ('SideVol', 'Polarizability'), ('SideVol', 'SASA'), ('SideVol', 'NCI'), ('SideVol', 'Mass'), ('Polarity', 'Polarizability'), ('Polarity', 'SASA'), ('Polarity', 'NCI'), ('Polarity', 'Mass'), ('Polarizability', 'SASA'), ('Polarizability', 'NCI'), ('Polarizability', 'Mass'), ('SASA', 'NCI'), ('SASA', 'Mass'), ('NCI', 'Mass'), ('HydroPho', 'None'), ('HydroPhIl', 'None'), ('Hbond', 'None'), ('SideVol', 'None'), ('Polarity', 'None'), ('Polarizability', 'None'), ('SASA', 'None'), ('NCI', 'None'), ('Mass', 'None')
```python
sequence="MALIPDLAMETWLLLAVSLVLLYL"
Vector45D=PCDTW45DConvert(sequence, normalize=True, NormType='MinMax')
print(Vector45D)
```
8)To find regions of conservation among a group of proteins stored in a FASTA file:
```python
FASTALoc="Your File Location"
display(FindConservation(FASTALoc))
```
This function outputs plotted data and returns a dataframe summarizing the mean and MAD distances for each pair of physicochemical properties. The data in the dataframe is most useful in making a broad determination of which properties are most conserved across the whole sequence and within the group of proteins being scrutinized. The plotted data is most useful in determining regions within the proteins that are conserved with respect to certain amino acid properties. For example, in the image below Hydrophilicity and Mass are well conserved from residues 8-26, 193-199 and from 203-209.
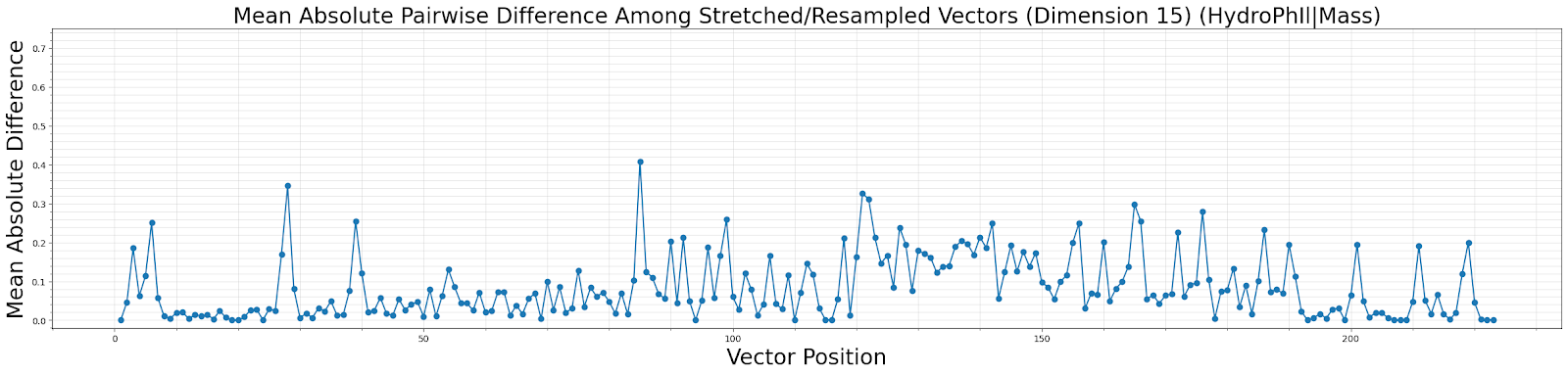
Dependency Citations:
Bio.Phylo:
Talevich, E., Invergo, B.M., Cock, P.J.A., & Chapman, B.A. (2012). Bio.Phylo: A unified toolkit for processing, analyzing, and visualizing phylogenetic trees in Biopython. BMC Bioinformatics, 13, 209
Biopython:
Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422–1423. https://doi.org/10.1093/bioinformatics/btp163
dtaidistance:
Wannes Meert, Kilian Hendrickx, Toon Van Craenendonck, Pieter Robberechts, Hendrik Blockeel, & Jesse Davis. (2022). DTAIDistance (Version v2). Zenodo. http://doi.org/10.5281/zenodo.5901139
Matplotlib:
Hunter, J. D. (2007). Matplotlib: A 2D graphics environment. Computing in Science & Engineering, 9(3), 90–95.
numpy:
Harris, C.R., Millman, K.J., van der Walt, S.J. et al. (2020). Array programming with NumPy. Nature 585, 357–362. DOI: 10.1038/s41586-020-2649-2.
pandas:
McKinney, W. (2010). Data Structures for Statistical Computing in Python. Proceedings of the 9th Python in Science Conference (SciPy 2010).
SciPy:
Pauli Virtanen, Ralf Gommers, Travis E. Oliphant, Matt Haberland, Tyler Reddy, David Cournapeau, Evgeni Burovski, Pearu Peterson, Warren Weckesser, Jonathan Bright, Stéfan J. van der Walt, Matthew Brett, Joshua Wilson, K. Jarrod Millman, Nikolay Mayorov, Andrew R. J. Nelson, Eric Jones, Robert Kern, Eric Larson, CJ Carey, İlhan Polat, Yu Feng, Eric W. Moore, Jake VanderPlas, Denis Laxalde, Josef Perktold, Robert Cimrman, Ian Henriksen, E.A. Quintero, Charles R Harris, Anne M. Archibald, Antônio H. Ribeiro, Fabian Pedregosa, Paul van Mulbregt, and SciPy 1.0 Contributors. (2020) SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nature Methods, 17(3), 261-272.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file pcdtw-0.4.1.tar.gz.
File metadata
- Download URL: pcdtw-0.4.1.tar.gz
- Upload date:
- Size: 14.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e4349edebbd20ca672cc36d7983961ddaae4d440582e90d5e21d74d870fd0537
|
|
| MD5 |
09838a45888b56ba9548b264f4edeb3d
|
|
| BLAKE2b-256 |
77635703e9bcf1a67e6199b3e7a2e8e466a9c3d26aa5892a8be7557bc2a6d0b9
|
File details
Details for the file pcdtw-0.4.1-py3-none-any.whl.
File metadata
- Download URL: pcdtw-0.4.1-py3-none-any.whl
- Upload date:
- Size: 15.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c9d8932e1394e1796a0c46cc02770ca8e057f8d892734c2be85329f1bee72f2d
|
|
| MD5 |
c796f0b1da6e0a27e77f43d27ce47536
|
|
| BLAKE2b-256 |
3f89daba35fc007cb97cc1118f45fdb33035dc41d955ae4960c6a811c00f6530
|







