Analyze centromere variation from short-read data using rare k-mers in alpha satellite sequences
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description

Maruyama cairn at Mt. Karamatsu
ascairn



ascairn (alpha-satellite cairn) is software for estimating centromere variation from short-read sequencing data using rare k-mers within centromere sequences.
For each chromosome, ascairn identifies the most likely pair of active alpha satellite higher-order repeat (HOR) haplogroups and the nearest proxy haplotypes from a reference panel (Shiraishi et al., bioRxiv, 2025).
ascairn accepts BAM/CRAM files aligned to either GRCh38 (hg38) or T2T-CHM13 (chm13). Centromere region BED files for each alignment reference are included in the resource repository.
Background
Human centromeres are composed largely of chromosome-specific alpha satellite higher-order repeat (HOR) arrays. The active alpha satellite HOR arrays (aHOR arrays), which are associated with CENP-A and kinetochore formation, show extensive sequence and structural variation among individuals. Because aHOR arrays are long and highly repetitive, they have historically been difficult to analyze with conventional short-read sequencing.
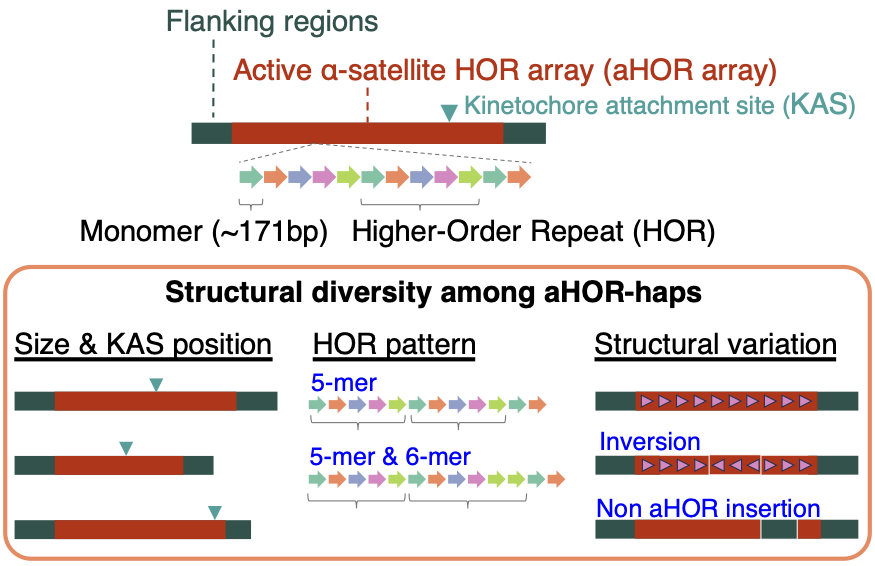
aHOR-haps comprise the aHOR array and flanking regions, and vary in size, KAS position, HOR pattern, and structural variants.
Long-read assemblies have recently revealed many complete centromeric haplotypes. However, applying long-read sequencing to thousands of population-scale or clinical samples remains costly and often impractical. ascairn addresses this gap by using rare k-mers within centromeric alpha satellite arrays as short-read-detectable markers of centromere haplotype structure.
What is a centromere haplogroup?
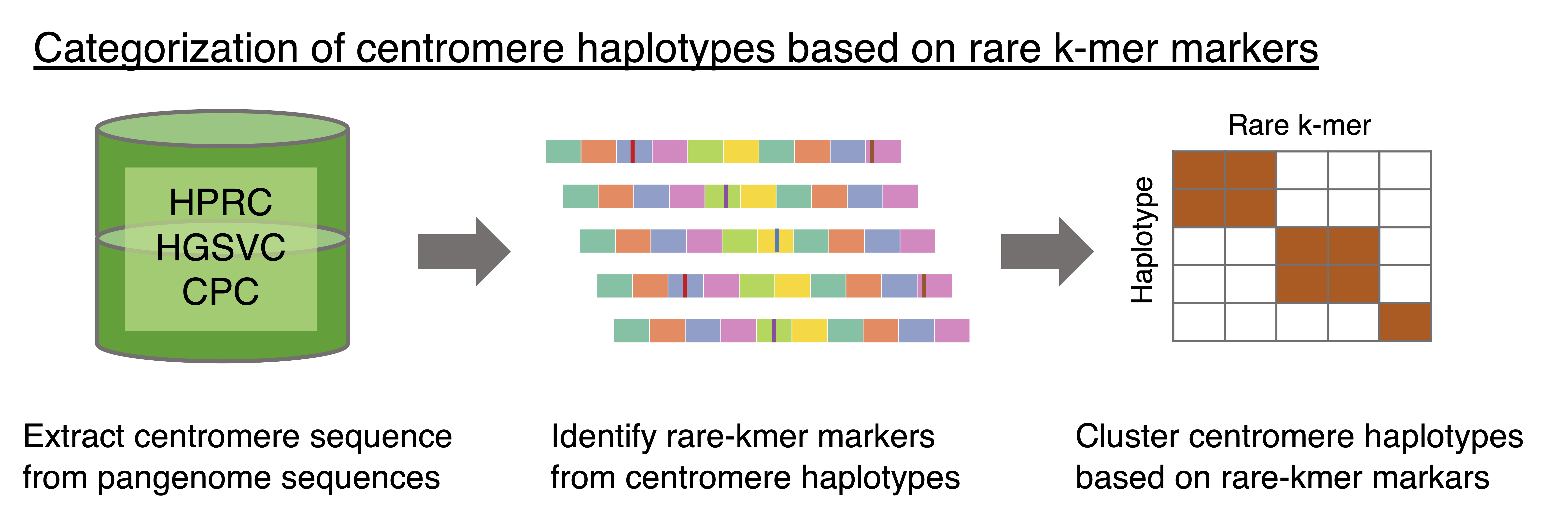
In ascairn, a centromere haplogroup is a chromosome-specific cluster of active alpha satellite HOR haplotypes (aHOR-haps) that share similar rare k-mer profiles, which serve as lineage-specific markers. These haplogroups are inferred from a reference panel of assembled aHOR-haps (the reference aHOR-hap panel) and often correspond to evolutionarily related centromere lineages with distinct structural features, such as differences in HOR organization, array size, or large structural variants.
Centromere haplogroups are assigned separately for each chromosome, describing variation at each individual centromere.
For more background, see Centromere haplogroups.
What ascairn does
ascairn infers aHOR-HGs from short-read whole-genome sequencing data. It extracts reads aligned to centromeric alpha satellite regions, generates a predefined rare k-mer count profile, and uses a probabilistic model to identify the most likely aHOR-HG pair for each chromosome. It also reports the closest proxy aHOR-hap pair from the reference aHOR-hap panel.
The reference aHOR-hap panel was constructed in advance by extracting centromeric sequences from publicly available long-read assemblies (HPRC, HGSVC, T2T Consortium) and is distributed through the ascairn_resource repository.
The main outputs are:
- the best-matching aHOR-HG (cluster) pair,
- the nearest proxy aHOR-hap pair,
- marker-level probability tables supporting the assignment.
ascairn does not assemble centromeres de novo. Its results depend on sequencing depth, read alignment quality, and the representation of related aHOR-haps in the supplied reference panel.
For more on the applications of ascairn, see Applications.
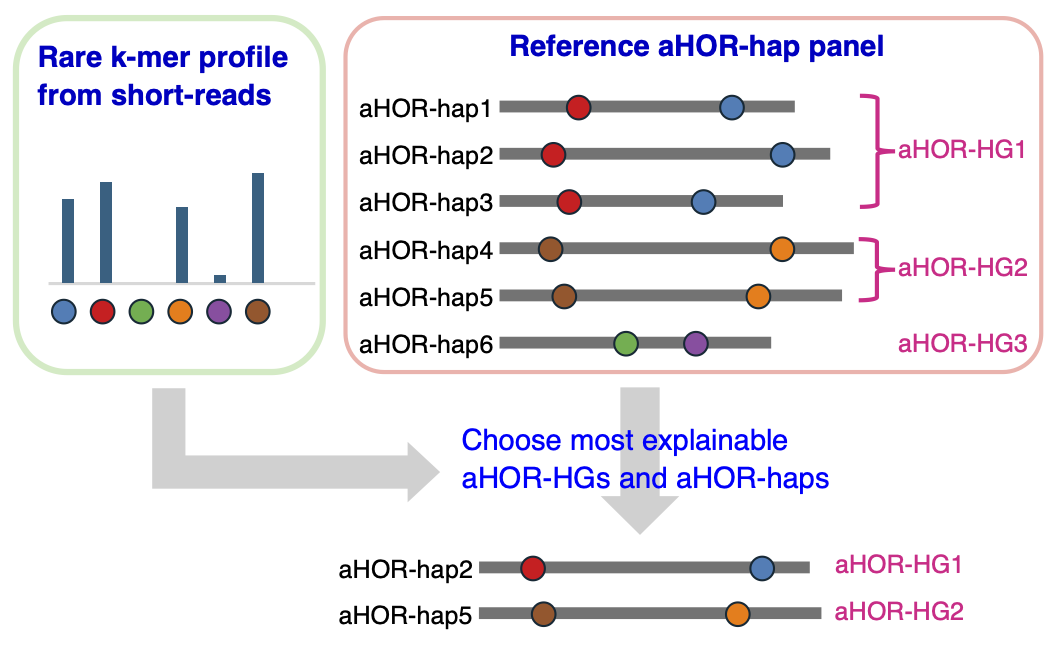
Overview of the ascairn workflow. Short-read WGS data are processed to count rare k-mers in centromeric regions; a probabilistic model is then used to infer the most likely aHOR-HG pair and the nearest proxy aHOR-hap pair from the reference panel.
Prerequisites
Software
Python packages
- click
- scipy
- polars
- boto3 (required only for accessing CRAM files in Amazon S3)
Installation
-
Install prerequisite software and ensure they are accessible via your
PATH. -
Install
ascairnfrom PyPI.
pip install ascairn
To enable access to CRAM files on Amazon S3, install with the s3 extra:
pip install ascairn[s3]
Or, to install from source (for development):
git clone https://github.com/friend1ws/ascairn.git
cd ascairn
pip install -e .
- Download ascairn resource files.
This repository contains the reference data required by ascairn, including:
- Rare k-mer list for alpha satellite sequences (
rare_kmer_list.fa) - Centromere region BED files for hg38 and chm13
- Per-chromosome k-mer information (
kmer_info/) - Per-chromosome haplotype-to-cluster mapping (
hap_info/)
- Rare k-mer list for alpha satellite sequences (
git clone https://github.com/friend1ws/ascairn_resource.git
After installation, your directory structure should look like this:
ascairn/
ascairn_resource/
└── resource/
├── common/
│ ├── cen_region_curated_margin_hg38.bed
│ ├── cen_region_curated_margin_chm13.bed
│ ├── chr22_long_arm_hg38.bed
│ ├── chr22_long_arm_chm13.bed
│ ├── chrX_short_arm_hg38.bed
│ └── chrX_short_arm_chm13.bed
└── panel/
└── ascairn_paper_2025/
├── rare_kmer_list.fa
├── kmer_info/
│ ├── chr1.kmer_info.txt.gz
│ ├── ...
│ └── chrX.kmer_info.txt.gz
└── hap_info/
├── chr1.hap_info.txt
├── ...
└── chrX.hap_info.txt
Quick Start
1. Prepare the sequence data
We use NA12877 from the 1000 Genomes Project, whose GRCh38-aligned CRAM is publicly available on AWS S3 and FTP.
Option 1: Direct S3 path (no download required)
If samtools is properly installed with S3 support, ascairn can read CRAM files directly from S3 without downloading. The file is:
s3://1000genomes/1000G_2504_high_coverage/additional_698_related/data/ERR3989340/NA12877.final.cram
Option 2: Download locally
Either via AWS CLI (public access, no AWS credentials required):
aws s3 cp --no-sign-request s3://1000genomes/1000G_2504_high_coverage/additional_698_related/data/ERR3989340/NA12877.final.cram seq_data/
aws s3 cp --no-sign-request s3://1000genomes/1000G_2504_high_coverage/additional_698_related/data/ERR3989340/NA12877.final.cram.crai seq_data/
Or via FTP:
wget ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989340/NA12877.final.cram -P seq_data/
wget ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3989340/NA12877.final.cram.crai -P seq_data/
2. Run the workflow
The simplest way to run ascairn is via type_all, which executes all steps (check_depth, kmer_count, cen_type) for chr1-22, chrX, and chrY (males only) in a single command.
Using Option 1 (direct S3 path):
ascairn type_all \
s3://1000genomes/1000G_2504_high_coverage/additional_698_related/data/ERR3989340/NA12877.final.cram \
-o output/NA12877 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference hg38 \
-t 8
Using Option 2 (downloaded CRAM):
ascairn type_all \
seq_data/NA12877.final.cram \
-o output/NA12877 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference hg38 \
-t 8
CRAM with a local reference fasta:
If the CRAM header's UR/MD5 cannot be resolved (network-isolated nodes, locally produced CRAMs, etc.), pass the reference fasta with -r/--reference_fasta. This is forwarded to samtools/mosdepth and works for check_depth, kmer_count, and type_all:
ascairn type_all \
seq_data/NA12877.final.cram \
-o output/NA12877 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference hg38 \
-r /path/to/GRCh38.fasta \
-t 8
3. Check the results
After successful execution, the main output file is:
output/NA12877.cen_type_all.txt
See Output Format for how to interpret the results.
Running with CHM13-aligned data
For users with CHM13-aligned data, here is an equivalent example using NA12878, whose CHM13-aligned CRAM is publicly available at the DDBJ mirror.
Option 1: Direct HTTPS URL (no download required)
If samtools is properly installed with libcurl support, ascairn can read CRAM files directly from HTTPS URLs:
ascairn type_all \
https://ddbj.nig.ac.jp/public/public-human-genomes/CHM13/1000Genomes/CRAM/NA12878/NA12878.cram \
-o output/NA12878 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference chm13 \
-t 8
Option 2: Download locally
wget https://ddbj.nig.ac.jp/public/public-human-genomes/CHM13/1000Genomes/CRAM/NA12878/NA12878.cram -P seq_data/
wget https://ddbj.nig.ac.jp/public/public-human-genomes/CHM13/1000Genomes/CRAM/NA12878/NA12878.cram.crai -P seq_data/
ascairn type_all \
seq_data/NA12878.cram \
-o output/NA12878 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference chm13 \
-t 8
The only differences from the GRCh38 workflow are (i) the input CRAM aligned to CHM13 and (ii) --reference chm13 in the command.
Commands
type_all
Runs the full workflow (check_depth → kmer_count → cen_type for all chromosomes) in a single command. Automatically determines biological sex and applies single-haplotype mode for chrX in males.
ascairn type_all \
seq_data/NA12877.final.cram \
-o output/NA12877 \
--resource_dir ascairn_resource/resource/panel/ascairn_paper_2025 \
--reference hg38 \
-t 8
| Option | Description | Required |
|---|---|---|
BAM_FILE |
Path to BAM or CRAM file (positional argument) | Yes |
-o |
Output path prefix | Yes |
--resource_dir |
Path to panel resource directory | Yes |
--reference |
Reference genome build (hg38 or chm13) |
Yes |
-t |
Number of threads | No (default: 8) |
Individual commands
The workflow consists of three commands that are normally run in order. You can run them individually for more control.
check_depth
Checks sequence coverage in a reference region (chr22 long arm) and determines biological sex by comparing chrX coverage.
ascairn check_depth \
seq_data/NA12877.final.cram \
-o output/NA12877.depth.txt \
--baseline_region ascairn_resource/resource/common/chr22_long_arm_hg38.bed \
--x_region ascairn_resource/resource/common/chrX_short_arm_hg38.bed \
-t 8
Output (output/NA12877.depth.txt):
Coverage: 37.24
Baseline region file: ascairn_resource/resource/common/chr22_long_arm_hg38.bed
Sex: male
ChrX region file: ascairn_resource/resource/common/chrX_short_arm_hg38.bed
ChrX coverage: 18.0
kmer_count
Extracts reads aligned to alpha satellite regions and counts occurrences of predefined rare k-mers.
ascairn kmer_count \
seq_data/NA12877.final.cram \
-o output/NA12877.kmer_count.txt \
--kmer_file ascairn_resource/resource/panel/ascairn_paper_2025/rare_kmer_list.fa \
--cen_region ascairn_resource/resource/common/cen_region_curated_margin_hg38.bed \
-t 8
Output (output/NA12877.kmer_count.txt): a two-column TSV with k-mer sequences and their counts.
cen_type
Identifies the most likely centromere cluster pair and nearest haplotype pair for a given chromosome. Requires the depth file from check_depth output.
ascairn cen_type \
output/NA12877.kmer_count.txt \
-o output/NA12877.chr22 \
--kmer_info ascairn_resource/resource/panel/ascairn_paper_2025/kmer_info/chr22.kmer_info.txt.gz \
--hap_info ascairn_resource/resource/panel/ascairn_paper_2025/hap_info/chr22.hap_info.txt \
--depth_file output/NA12877.depth.txt
For male samples on chrX, add the --single_hap option (type_all handles this automatically based on check_depth output).
Output files (using output/NA12877.chr22 as the output prefix):
| File | Description |
|---|---|
*.cen_type.txt |
Best cluster pair, haplotype pair, and hap_info annotations (e.g., Contig_len) |
*.cluster.hap_pair.txt |
Ranked cluster pairs with log-likelihoods. The first data row is the best pair. |
*.haplotype.hap_pair.txt |
Ranked haplotype pairs with log-likelihoods. The first data row is the best pair. |
*.cluster.marker_prob.txt |
Per-marker copy number probabilities used for cluster assignment |
*.haplotype.marker_prob.txt |
Per-marker copy number probabilities used for haplotype assignment |
Output Format
cen_type_all.txt
The main result file (generated by type_all) is a TSV with one row per chromosome:
| Column | Description |
|---|---|
| Chr | Chromosome (chr1-chr22, chrX, chrY) |
| Cluster_1 | Best-matching cluster ID for the first allele |
| Cluster_2 | Best-matching cluster ID for the second allele |
| Haplotype_1 | Nearest haplotype ID for the first allele (e.g., HG00268.mat) |
| Haplotype_2 | Nearest haplotype ID for the second allele |
| additional columns | Any extra columns from hap_info.txt are automatically appended with _1/_2 suffixes (e.g., Contig_len_1, Contig_len_2) |
For male chrX, _2 columns are NA.
Per-chromosome cen_type.txt
Each cen_type run produces a *.cen_type.txt file with the same columns as above (without the Chr column). The cen_type_all.txt file is the concatenation of these per-chromosome files.
Cluster and haplotype pair files
These files list all candidate pairs ranked by log-likelihood (higher = better fit):
| Column | Description |
|---|---|
| Cluster1 / Haplotype1 | First member of the pair |
| Cluster2 / Haplotype2 | Second member of the pair |
| Loglikelihood | Log-likelihood of the pair given the observed k-mer counts |
Performance
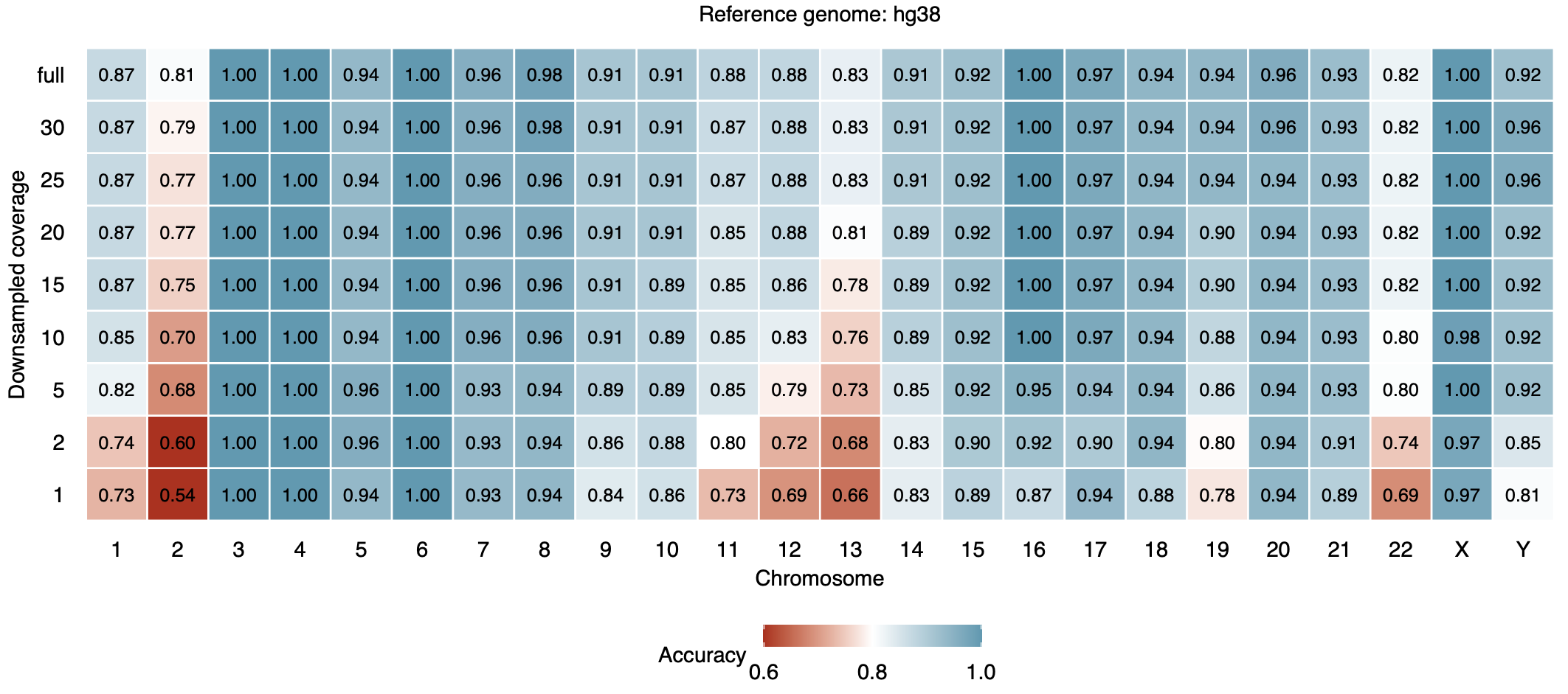
We evaluated ascairn using leave-one-individual-out cross-validation on the reference panel: for each individual, both parental aHOR-haps were removed from the panel, and the resulting model was used to infer the haplogroup pair from the individual's short-read WGS data. To assess robustness to sequencing depth, each sample was downsampled to 1–30x coverage.
The figures below show haplogroup pair assignment accuracy for each chromosome at various downsampled coverages, for short-read WGS data aligned to GRCh38 (upper) and CHM13 v2.0 (lower):
GRCh38-aligned data:
CHM13-aligned data:
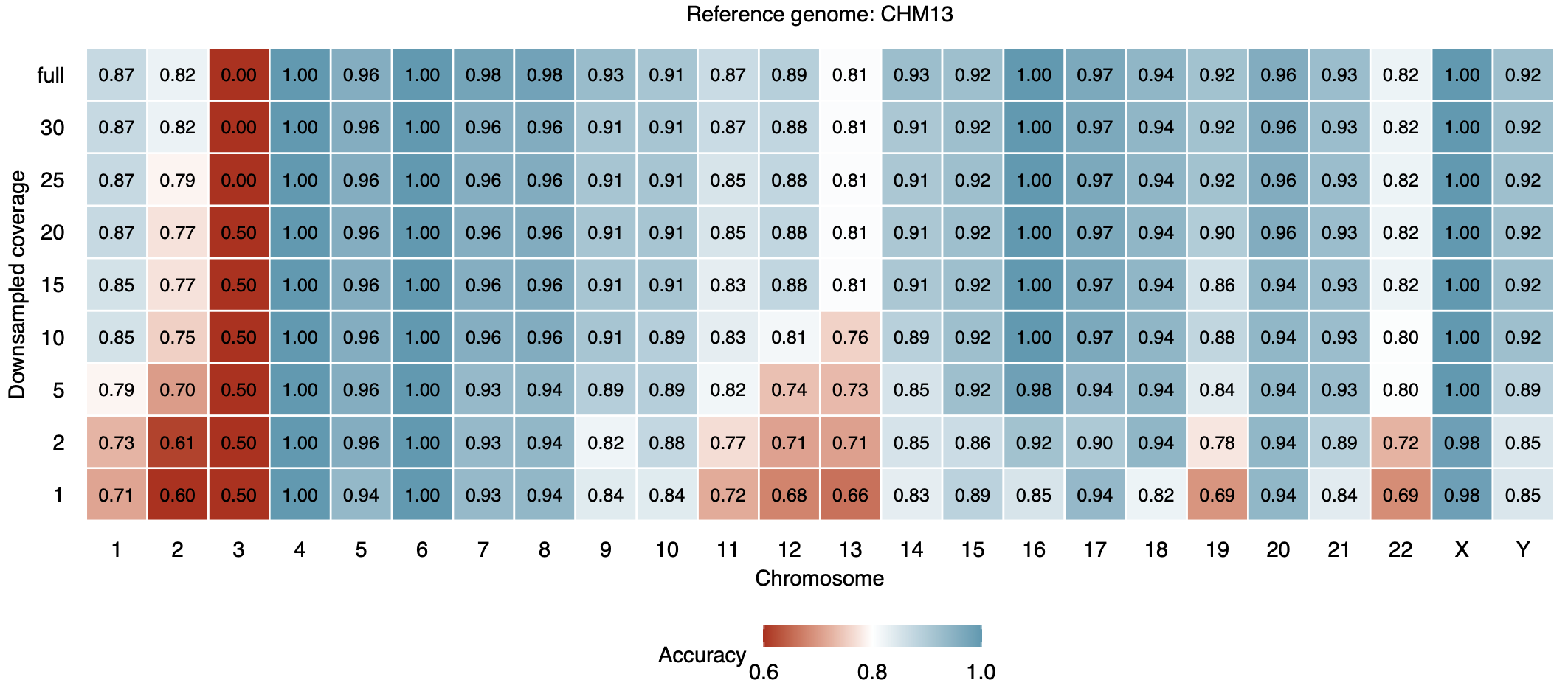
Accuracy is generally high (>90%) for most chromosomes at coverage ≥ 5x, with comparable performance between GRCh38- and CHM13-aligned data. See Shiraishi et al., bioRxiv, 2025 for details.
Notes
- chrY: chrY is processed only for male samples (determined automatically by
check_depthfrom the chrX coverage ratio). - Test data: The Quick Start example uses NA12877 from the 1000 Genomes Project high-coverage dataset. This sample was chosen because it is publicly accessible via both AWS S3 and FTP, enabling reproducible testing without restricted data access.
Citation
Shiraishi Y, Ochi Y, Sugawa M, Sakamoto Y, Kimura K, Tsujimura T, Okada A, Okuda R, Namba S, Miyauchi T, Mateos RN, Suzuki H, Chiba K, Ito Y, Nakamura W, Ohka F, Motomura K, Yamamoto T, Kawai Y, Okada Y, Suzuki H, Kato M, Saito R, Garrison E, Logsdon GA, Ogawa S. Rare k-mers reveal centromere haplogroups underlying human diversity and cancer translocations. bioRxiv. 2025. doi: 10.1101/2025.07.26.666712
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ascairn-0.1.2.tar.gz.
File metadata
- Download URL: ascairn-0.1.2.tar.gz
- Upload date:
- Size: 37.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c4f929cfabd6fdb2ef9d4f664790e05a40f6de84f45739885819900615ac0c53
|
|
| MD5 |
f5f534c11933ebafcc91f4ca6ac253ef
|
|
| BLAKE2b-256 |
f810c0fb61f31ff74e6c40c9c7114d434796da9816c0a299db67ac6f248b15d1
|
Provenance
The following attestation bundles were made for ascairn-0.1.2.tar.gz:
Publisher:
python-publish.yml on friend1ws/ascairn
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ascairn-0.1.2.tar.gz -
Subject digest:
c4f929cfabd6fdb2ef9d4f664790e05a40f6de84f45739885819900615ac0c53 - Sigstore transparency entry: 1665647694
- Sigstore integration time:
-
Permalink:
friend1ws/ascairn@3eb50bdd8865f3e2a6a20907cf1c190aa71d4d33 -
Branch / Tag:
refs/tags/v0.1.2 - Owner: https://github.com/friend1ws
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
python-publish.yml@3eb50bdd8865f3e2a6a20907cf1c190aa71d4d33 -
Trigger Event:
release
-
Statement type:
File details
Details for the file ascairn-0.1.2-py3-none-any.whl.
File metadata
- Download URL: ascairn-0.1.2-py3-none-any.whl
- Upload date:
- Size: 33.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1751bcaf71643c78eb9c0927a0afee63bcb042c1ff420ecfd444e8efa50b3bc7
|
|
| MD5 |
e90feb4b246a0c67073e7eec5b3333e7
|
|
| BLAKE2b-256 |
df6c9994e8a8190edce653c13452e0c0356ffef7ff04e9b436154d3a01e83f85
|
Provenance
The following attestation bundles were made for ascairn-0.1.2-py3-none-any.whl:
Publisher:
python-publish.yml on friend1ws/ascairn
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ascairn-0.1.2-py3-none-any.whl -
Subject digest:
1751bcaf71643c78eb9c0927a0afee63bcb042c1ff420ecfd444e8efa50b3bc7 - Sigstore transparency entry: 1665647828
- Sigstore integration time:
-
Permalink:
friend1ws/ascairn@3eb50bdd8865f3e2a6a20907cf1c190aa71d4d33 -
Branch / Tag:
refs/tags/v0.1.2 - Owner: https://github.com/friend1ws
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
python-publish.yml@3eb50bdd8865f3e2a6a20907cf1c190aa71d4d33 -
Trigger Event:
release
-
Statement type:







