CheckV: Assessing the quality of metagenome-assembled viral genomes


Project description




CheckV is a fully automated command-line pipeline for assessing the quality of single-contig viral genomes, including identification of host contamination for integrated proviruses, estimating completeness for genome fragments, and identification of closed genomes.
Quick links
Quickstart
Installation
CheckV database
Running CheckV
How it works
Main output files
Known issues
Frequently asked questions
Supporting scripts
Citation
Quickstart
Run the following commands to install CheckV, download the database, and run the program.
Replace </path/to/database> and <input_file.fna> with the correct file paths:
conda install -c conda-forge -c bioconda checkv
checkv download_database ./
export CHECKVDB=</path/to/database>
checkv end_to_end <input_file.fna> output_directory -t 16
Installation
There are three methods to install CheckV:
-
Using conda or mamba (recommended):
conda install -c conda-forge -c bioconda checkv=1.0.1 -
Using pip:
pip install checkv -
Using docker: see section below
If you decide to install CheckV via pip, make sure you also have the following external dependencies installed:
- DIAMOND (v2.1.8): https://github.com/bbuchfink/diamond
- HMMER (v3.3): http://hmmer.org/
- Prodigal-gv (v2.6.3): https://github.com/apcamargo/prodigal-gv
The versions listed above were the ones that were properly tested. There is a known issue with DIAMOND v2.1.9 which should be avoided.
If you decide to install CheckV via docker, note that the docker image may not represent the latest release. Here are the commands to pull and run the image:
docker pull antoniopcamargo/checkv
docker run -ti --rm -v "$(pwd):/app" antoniopcamargo/checkv end_to_end input_file.fna output_directory -t 16
CheckV database
If you install using conda or pip you will need to download the database:
checkv download_database ./
You'll need to update your environment or use the -d flag to specify the CHECKVDB location:
export CHECKVDB=/path/to/checkv-db
Some users may wish to update the database using their own complete genomes:
checkv update_database /path/to/checkv-db /path/to/updated-checkv-db genomes.fna
Some users may wish to download a specific database version. See https://portal.nersc.gov/CheckV/ for an archive of all previous database version. If you go this route then you'll need to build the DIAMOND database manually:
wget https://portal.nersc.gov/CheckV/checkv-db-archived-version.tar.gz
tar -zxvf checkv-db-archived-version.tar.gz
cd /path/to/checkv-db/genome_db
diamond makedb --in checkv_reps.faa --db checkv_reps
The database is frequently updated. You can keep track of those updates here:
https://portal.nersc.gov/CheckV/README.txt
Running CheckV
There are two ways to run CheckV:
Using a single command to run the full pipeline (recommended):
checkv end_to_end input_file.fna output_directory -t 16
Using individual commands for each step in the pipeline:
checkv contamination input_file.fna output_directory -t 16
checkv completeness input_file.fna output_directory -t 16
checkv complete_genomes input_file.fna output_directory
checkv quality_summary input_file.fna output_directory
For a full listing of checkv programs and options, use: checkv -h and checkv <program> -h
Known issues
- There is an issue with DIAMOND v2.1.9 that causes a core dump
- For 0.9.0 you may get an error that some prodigal tasks failed.
- For >=0.9.0 if you get error that the diamond database was not found re-download the database using
checkv download_database - For v0.8.1 sometimes conda installed an older version of DIAMOND causing an error. Make sure conda has installed DIAMOND version >= 2.0.9
How it works
The pipeline can be broken down into 4 main steps:
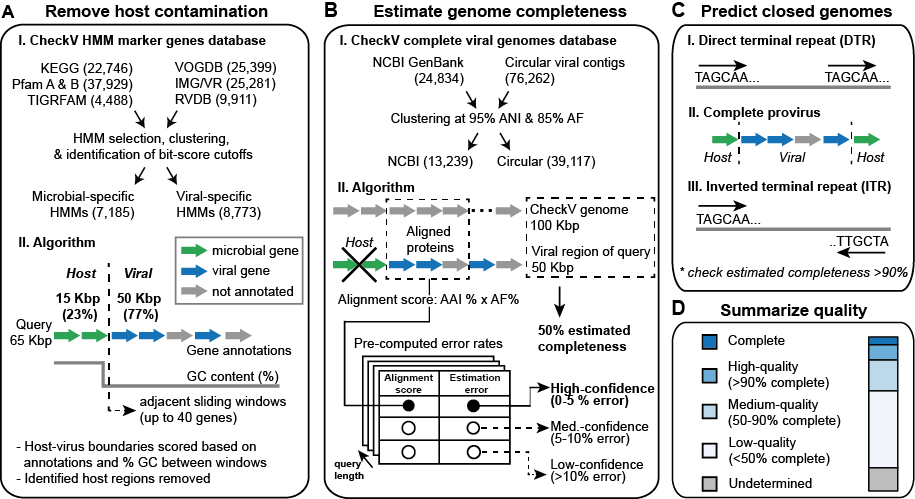
A: Remove host contamination on proviruses
- Genes are first annotated as viral or microbial based on comparison to a custom database of HMMs
- CheckV scans over the contig (5' to 3') comparing gene annotations and GC content between a pair of adjacent gene windows
- This information is used to compute a score at each intergenic position and identify host-virus breakpoints
- Works best for contigs that are mostly viral
B: Estimate genome completeness
- Proteins are first compared to the CheckV genome database using AAI (average amino acid identity)
- After identifying the top hits, completeness is computed as a ratio between the contig length (or viral region length for proviruses) and the length of matched reference
- A confidence level is reported based on the strength of the alignment
- Generally, high- and medium-confidence estimates are quite accurate
- Less frequently, your viral genome may not have a close match to the CheckV database; in these cases CheckV estimates the completeness based on the viral HMMs identified on the contig
- Based on the HMMs found, CheckV returns the estimated range for genome completeness (e.g. 35% to 60% completeness), which represents the 90% confidence interval based on the distribution of lengths of reference genomes with the same viral HMMs
C: Predict closed genomes
-
Direct terminal repeats (DTRs)
- Repeated sequence of >20-bp at start/end of contig
- Most trusted signature in our experience
- May indicate circular genome or linear genome replicated from a circular template (i.e. concatamer)
-
Proviruses
- Viral region with predicted host boundaries at 5' and 3' ends (see panel A)
- Note: CheckV will not detect proviruses if host regions have already been removed (e.g. using VIBRANT or VirSorter)
-
Inverted terminal repeats (ITRs)
- Repeated sequence of >20-bp at start/end of contig (3' repeat is inverted)
- Least trusted signature
-
For all the methods above, CheckV also checks whether the contig is approximately the correct sequence length based on estimated completeness; this is important because terminal repeats can represent artifacts of metagenomic assembly
D: Summarize quality.
Based on the results of A-C, CheckV generates a report file and assigns query contigs to one of five quality tiers (consistent with and expand upon the MIUViG quality tiers):
- Complete (see panel C)
- High-quality (>90% completeness)
- Medium-quality (50-90% completeness)
- Low-quality (<50% completeness)
- Undetermined quality
Output files
quality_summary.tsv
This contains integrated results from the three main modules and should be the main output referred to. Below is an example to demonstrate the type of results you can expect in your data:
| contig_id | contig_length | provirus | proviral_length | gene_count | viral_genes | host_genes | checkv_quality | miuvig_quality | completeness | completeness_method | complete_genome_type | contamination | kmer_freq | warnings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5325 | No | NA | 11 | 0 | 2 | Not-determined | Genome-fragment | NA | NA | NA | 0 | 1 | no viral genes detected |
| 2 | 41803 | No | NA | 72 | 27 | 1 | Low-quality | Genome-fragment | 21.99 | AAI-based (medium-confidence) | NA | 0 | 1 | flagged DTR |
| 3 | 38254 | Yes | 36072 | 54 | 23 | 2 | Medium-quality | Genome-fragment | 80.3 | HMM-based (lower-bound) | NA | 5.7 | 1 | |
| 4 | 67622 | No | NA | 143 | 25 | 0 | High-quality | High-quality | 100 | AAI-based (high-confidence) | NA | 0 | 1.76 | high kmer_freq |
| 5 | 98051 | No | NA | 158 | 27 | 1 | Complete | High-quality | 100 | AAI-based (high-confidence) | DTR | 0 | 1 |
In the example, above there are results for 6 viral contigs:
- The first 5325 bp contig has no completeness prediction, which is indicated by 'Not-determined' for the 'checkv_quality' field. This contig also has no viral genes identified, so there's a chance it may not even be a virus.
- The second 41803 bp contig is classified as 'Low-quality' since its completeness is <50%. This is estimate is based on the 'AAI' method. Note that only either high- or medium-confidence estimates are reported in the quality_summary.tsv file. You can see 'completeness.tsv' for more details. This contig had a DTR, but it was flagged for some reason (see complete_genomes.tsv for details)
- The third contig is considered 'Medium-quality' since its completeness is estimated to be 80%, which is based on the 'HMM' method. This means that it was too novel to estimate completeness based on AAI, but shared an HMM with CheckV reference genomes. Note that this value represents a lower bound (meaning the true completeness may be higher but not lower than this value). Note that this contig is also classified as a provirus.
- The fourth contig is classified as High-quality based on a completness of >90%. However, note that value of 'kmer_freq' is 1.7. This indicates that the viral genome is represented multiple times in the contig. These cases are quite rare, but something to watch out for.
- The fifth contig is classified as Complete based on the presence of a direct terminal repeat (DTR) and has 100% completeness based on the AAI method. This sequence can condifently treated as a complete genome.
completeness.tsv
A detailed overview of how completeness was estimated:
| contig_id | contig_length | proviral_length | aai_expected_length | aai_completeness | aai_confidence | aai_error | aai_num_hits | aai_top_hit | aai_id | aai_af | hmm_completeness_lower | hmm_completeness_upper | hmm_hits |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9837 | 5713 | 53242.8 | 10.7 | high | 3.7 | 10 | DTR_517157 | 78.5 | 34.6 | 5 | 15 | 4 |
| 2 | 39498 | NA | 37309 | 100.0 | medium | 7.7 | 11 | DTR_357456 | 45.18 | 30.46 | 75 | 100 | 22 |
| 3 | 29224 | NA | 44960.1 | 65.8 | low | 15.2 | 17 | DTR_091230 | 39.74 | 19.54 | 52 | 70 | 10 |
| 4 | 23404 | NA | NA | NA | NA | NA | 0 | NA | NA | NA | NA | NA | 0 |
In the example, above there are results for 4 viral contigs:
- The first proviral contig has an estimated completeness of 10.7% using on the AAI-based method (100 x 5713 / 53242.8). The confidence for this estimate is high, based on a relative estimated error of 3.7%, which is in turn based on the aai_id (average amino acid identity) and aai_af (alignment fraction of contig) to the CheckV reference DTR_517517
- The second contig has a completeness of 100% using the AAI-based method and a completeness range of 75 - 100% using the HMM-based method. Note that the contig length is a bit longer than the expected genome length of 37,309 bp.
- The third contig is estimated to be 65.8% complete based on the AAI approach. However we can't trust this all that much since the aai_confidence is low (meaning the top hit based on AAI was fairly weak). To be conservative, we may wish to report the range of completeness (52-70%) based on the HMM approach
- The last contig doesn't have any hits based on AAI and doesn't have any viral HMMs, so there's nothing we can say about this sequence
contamination.tsv
A detailed overview of how contamination was estimated:
| contig_id | contig_length | total_genes | viral_genes | host_genes | provirus | proviral_length | host_length | region_types | region_lengths | region_coords_bp | region_coords_genes | region_viral_genes | region_host_genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 98051 | 158 | 27 | 1 | No | NA | NA | NA | NA | NA | NA | NA | NA |
| 2 | 38254 | 54 | 23 | 2 | Yes | 36072 | 2182 | host,viral | 1-2182,2183-38254 | 1-2182,2183-38254 | 1-4,5-54 | 0,23 | 2,0 |
| 3 | 6930 | 9 | 1 | 2 | Yes | 3023 | 3907 | viral,host | 3023,3907 | 1-3023,3024-6930 | 1-5,6-9 | 1,0 | 0,2 |
| 4 | 101630 | 103 | 7 | 24 | Yes | 28170 | 73460 | host,viral,host | 46804,28170,26656 | 1-46804,46805-74974,74975-101630 | 1-43,44-85,86-103 | 0,7,0 | 13,0,11 |
In the example, above there are results for 4 viral contigs:
- The first contig is not a predicted provirus
- The second contig has a predicted host region covering 2182 bp
- The third 6930 bp contig has a host region identified on the left side
- The fourth 101630 bp contig has 103 genes, including 7 viral and 24 host genes. CheckV identified two host-virus boundaries
complete_genomes.tsv
A detailed overview of putative complete genomes identified:
| contig_id | contig_length | prediction_type | confidence_level | confidence_reason | repeat_length | repeat_count |
|---|---|---|---|---|---|---|
| 1 | 44824 | DTR | high | AAI-based completeness > 90% | 253 | 2 |
| 2 | 38147 | DTR | low | Low complexity TR; Repetetive TR | 20 | 10 |
| 3 | 67622 | DTR | low | Multiple genome copies detected | 26857 | 2 |
| 4 | 5477 | ITR | medium | AAI-based completeness > 80% | 91 | 2 |
| 5 | 101630 | Provirus | not-determined | NA | NA | NA |
In the example, above there are results for 5 viral contigs:
- The first viral contig has a direct terminal repeat of 253 bp. It is classified as high-confidence based on having an estimated completeness > 90%
- The second viral contig has a 20-bp DTR, but is the DTR is low complexity and can't be trusted, resulting in a low confidence level. The DTR also occurs 10x and is considered repetetive.
- The third viral contig has DTR of 26857 bp! This indicates that a very large fraction of the genome is repeated. CheckV classifies these as low confidence, but users may which to manually resolve these duplications
- The fourth viral contig has ITR of 91 bp. This is considered medium-confidence based on having AAI-based completeness > 80%
- The fifth viral contig is flanked by host on both sides (provirus). However CheckV was unable to assess completeness, so the confidence is left as not-determined
Frequently asked questions
Q: Can I use CheckV for viral prediction? A: CheckV reports two types of viral signatures: (1) gene-level hits to viral or host HMMs, and (2) genome-level matches to viruses in the CheckV database. Both types of information are certainly useful for discriminating between viruses and non-viruses. However, for proper virus prediction, we recommend using geNomad: https://github.com/apcamargo/genomad
Q: What is the difference between AAI- and HMM-based completeness? A: AAI-based completeness produces a single estimated completeness value, which was designed to be very accurate and can be trusted when the reported confidence level is medium or high. HMM-based completeness gives the 90% confidence interval of completeness (e.g. 30-75%) in cases where AAI-based completeness is not reliable. In this example, we can be 90% sure (in theory) that the completeness is between 30% to 75%.
Q: What is the meaning of the kmer_freq field? A: This is a measure of how many times the viral genome is represented in the contig. Most times this is very close to 1.0. In rare cases assembly errors may occur in which the contig sequence represents multiple concatenated copies of the viral genome. In these cases genome_copies will exceed 1.0.
Q: Why does my DTR contig have <100% estimated completeness? A: If the estimated completeness is close to 100% (e.g. 90-110%) then the query is likely complete. However sometimes incomplete genome fragments may contain a direct terminal repeat (DTR), in which case we should expect their estimated completeness to be <90%, and sometimes much less. In other cases, the contig will truly be circular, but the estimated completeness is incorrect. This may also happen if the query a complete segment of a multipartite genome (common for RNA viruses). By default, CheckV uses the 90% completeness cutoff for verification, but a user may wish to make their own judgement in these ambiguous cases.
Q: Why is my sequence considered "high-quality" when it has high contamination? A: CheckV determines sequence quality solely based on completeness. Host contamination is easily removed, so is not factored into these quality tiers.
Q: I performed binning and generated viral MAGs. Can I use CheckV on these? A: CheckV can estimate completeness but not contamination for these. You'll need to concatentate the contigs from each MAG into a single sequence prior to running CheckV.
Q: Can I use CheckV to predict (pro)viruses from whole (meta)genomes? A: Possibly, though this has not been tested. Instead we recommend using geNomad: https://github.com/apcamargo/genomad
Q: How should I handle putative "closed genomes" with no completeless estimate? A: In some cases, you won't be able to verify the completeness of a sequence with terminal repeats or provirus integration sites. DTRs are a fairly reliable indicator (>90% of the time) and can likely be trusted with no completeness estimate. However, complete proviruses and ITRs are much less reliable indicators, and therefore require >90% estimated completeness.
Q: Why is my contig classified as "undetermined quality"? A: This happens when the sequence doesn't match any CheckV reference genome with high enough similarity to confidently estimate completeness and doesn't have any viral HMMs. There are a few explanations for this, in order of likely frequency: 1) your contig is very short, and by chance it does not share any genes with a CheckV reference, 2) your contig is from a very novel virus that is distantly related to all genomes in the CheckV database, 3) your contig is not a virus at all and so doesn't match any of the references.
Q: How should I handle sequences with "undetermined quality"? A: While it is not possible to estimate completeness for these, you may choose to still analyze sequences above a certain length (e.g. >30 kb).
Q: Why are sequences with 0 viral genes included in the CheckV output Currently, CheckV assumes that the input sequences represent viruses and attempts to estimate their quality. Some input sequences may be derived from bacteria, plasmids, or other sources, and may therefore have 0 viral genes detected.
Supporting code
Rapid genome clustering based on pairwise ANI
First, create a blast+ database:
makeblastdb -in <my_seqs.fna> -dbtype nucl -out <my_db>
Next, use megablast from blast+ package to perform all-vs-all blastn of sequences:
blastn -query <my_seqs.fna> -db <my_db> -outfmt '6 std qlen slen' -max_target_seqs 10000 -o <my_blast.tsv> -num_threads 32
Next, calculate pairwise ANI by combining local alignments between sequence pairs:
anicalc.py -i <my_blast.tsv> -o <my_ani.tsv>
Finally, perform UCLUST-like clustering using the MIUVIG recommended-parameters (95% ANI + 85% AF):
aniclust.py --fna <my_seqs.fna> --ani <my_ani.tsv> --out <my_clusters.tsv> --min_ani 95 --min_tcov 85 --min_qcov 0
The file <my_clusters.tsv> contains the clustering results. The first column is the cluster representative, and the second column contains cluster members.
The file <my_ani.tsv> contains the pairwise ANI results. The columns are:
- query_id: query identifier
- target_id: checkv reference genome identifier
- alignment_count: number of blastn alignments
- ani: average nucleotide identity
- query_coverage: percent of query genome covered by alignments
- target_coverage: percent of target genome covered by alignments
Citation
If you used the software in your research, please cite:
Nayfach, S., Camargo, A.P., Schulz, F. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol 39, 578–585 (2021). https://doi.org/10.1038/s41587-020-00774-7
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file checkv-1.0.3.tar.gz.
File metadata
- Download URL: checkv-1.0.3.tar.gz
- Upload date:
- Size: 925.9 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.10.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
2438516f270191267a9860dfe31bf596d64a4fbc16be922b46fb6a4fd98d762a
|
|
| MD5 |
5edadf73aa416ba3aa1e2e48384854f0
|
|
| BLAKE2b-256 |
95cbd4e309371f52541ff779ca1c275283b0f7e4afd2ec3b4dbe1d134df59549
|
File details
Details for the file checkv-1.0.3-py3-none-any.whl.
File metadata
- Download URL: checkv-1.0.3-py3-none-any.whl
- Upload date:
- Size: 34.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.0.0 CPython/3.10.14
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e09b331636042420527b40c74c3ec765dd17aecb4816cac8749f4d3e6ac77a21
|
|
| MD5 |
eb803755e56d142e10424776247ac8e2
|
|
| BLAKE2b-256 |
771d78f5748c088bea1cd7142d42745867c99eb852cefe3dd319c067ada664f9
|







