Terminal-first molecular dynamics viewer for GROMACS trajectories.
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
cmd
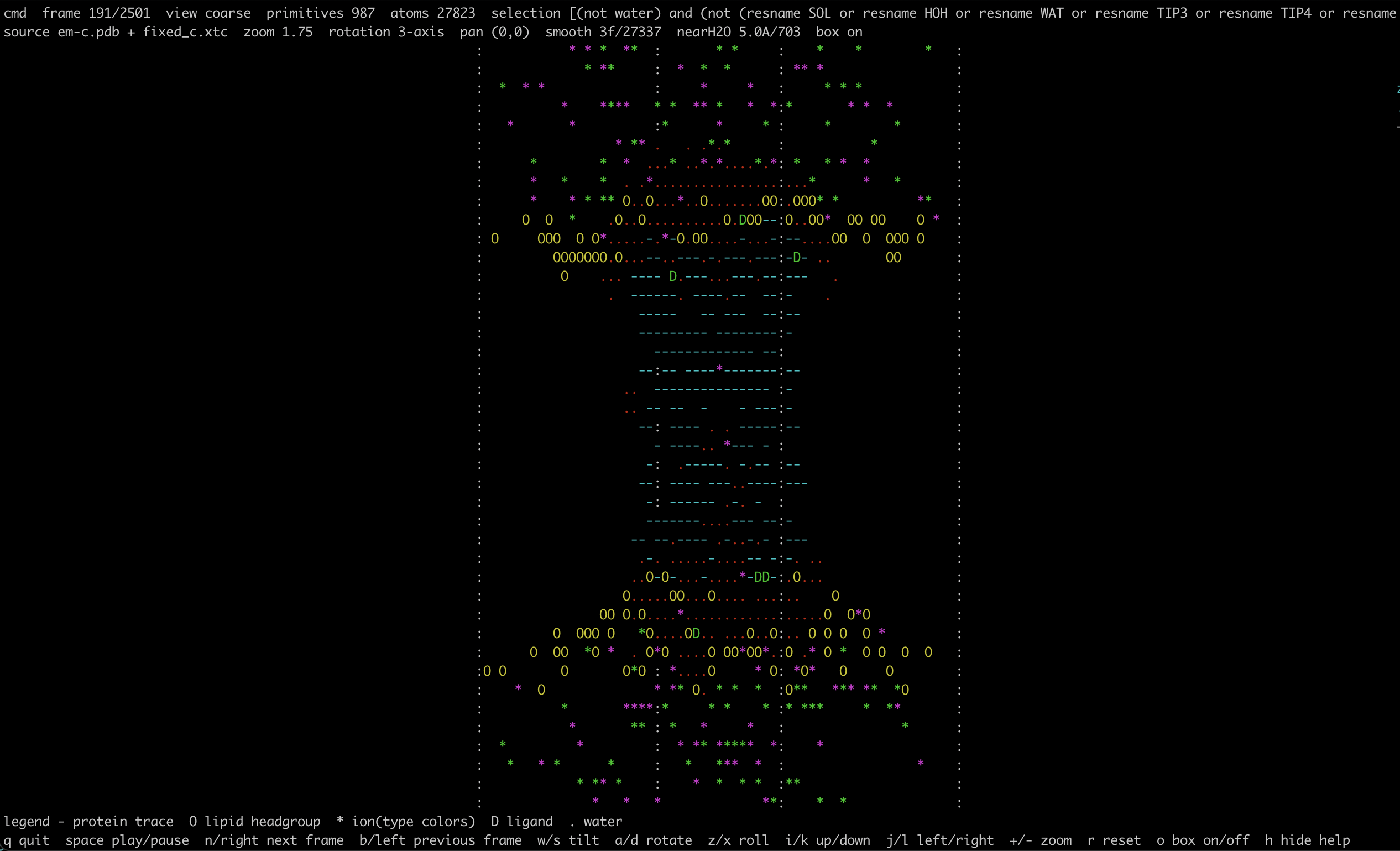
cmd is a terminal-first molecular dynamics viewer aimed at quick inspection of
simulation systems on local machines, HPC clusters, and remote shells. The
first prototype focuses on:
.pdb/.grostructures with optional.xtc/.dcdtrajectories- terminal-only rendering with no GUI dependencies
- water hidden by default
- atom selections that stay close to VMD workflows
- a single-command install path through
piporcondalater - responsive loading for quick trajectory sanity checks
- OpenMM-friendly
.pdb + .dcdworkflows in addition to GROMACS inputs
The internal Python package is named cmd_viewer to avoid colliding with
Python's standard-library cmd module. The installed executable remains cmd.
MVP design
The initial package is split into a few small layers:
cli.py: argument parsing and startuploader.py: MDAnalysis universe loading and frame accessselection.py: default visibility rules and VMD-like selection normalizationcamera.py: viewport state and 3D transformscolors.py: terminal palette and biomolecular class heuristicsrepresentations.py: mode-specific point, trace, and coarse primitivesrender.py: point-cloud projection into terminal cellsapp.py: curses event loop and playback controls
The rendering model is intentionally simple. The viewer supports a few startup
visualization modes selected with --view-mode:
points: atom-level point cloud for fast whole-system inspectiontrace: residue-level backbone-like trace usingCA/BB/P/C4'coarse: protein as dash traces, lipid headgroups asO, ions as colored*, and one coarse primitive for other non-protein residuescartoon: protein rendered as directional ASCII backbone segments, with the same coarse non-protein markers ascoarse, plus a simple secondary-structure heuristic that distinguishes helix-, sheet-, and loop-like segments
For large systems, the renderer downsamples to a bounded number of displayed point primitives so the viewer remains responsive over SSH and on shared systems. The viewer also draws a small XYZ axis triad so the current perspective remains legible, supports in-plane panning for zoomed inspection, and can overlay orthorhombic box boundaries when unit-cell dimensions are available.
Install
pip install .
For local iteration during development:
pip install -e .
Once published on PyPI:
pip install cmd-viewer
Release To PyPI
This repository includes a GitHub Actions workflow at
.github/workflows/release.yml that:
- builds an sdist and wheel
- runs
twine checkon the generated distributions - publishes to PyPI using Trusted Publishing when a GitHub release is published
- uses the GitHub Actions environment
pypifor an extra approval boundary
This repository also includes .github/workflows/package-checks.yml, which
builds the package, verifies version consistency, runs tests, and checks the
README/distributions on pushes, pull requests, and manual runs.
One-time PyPI setup:
- In PyPI, create a pending GitHub Actions publisher for project
cmd-viewer. - Point it at repository
Kopec-Lab/cmd-viewer. - Set the workflow file to
.github/workflows/release.yml. - Set the environment name to
pypi. - Make sure the GitHub release is created from the version you want to publish.
Typical release flow:
# update CHANGELOG.md if needed
# update version in pyproject.toml and src/cmd_viewer/__init__.py if needed
git tag v0.1.1
git push origin v0.1.1
Then publish a GitHub release for that tag. The workflow will build the package
and upload it to PyPI automatically after the pypi environment rules, if any,
are satisfied.
Once published, installation on a remote machine becomes:
pip install cmd-viewer
For the ongoing maintenance and release workflow, see
RELEASING.md.
Run
cmd system.gro traj.xtc
cmd openmm_system.pdb traj.dcd
cmd system.pdb --selection "protein or resname POPC"
cmd system.gro traj.xtc --show-water
cmd system.gro traj.xtc --view-mode trace --selection protein
cmd system.gro traj.xtc --view-mode coarse
cmd system.gro traj.xtc --selection lipids
cmd system.gro traj.xtc --selection "resname K" --color-mode resid
cmd system.gro traj.xtc --selection protein --near-water 5
cmd system.gro traj.xtc --view-mode cartoon --selection protein
cmd system.gro traj.xtc --view-mode coarse --selection "protein or resname POPC or resname CHOL or element K or element CL"
cmd system.gro traj.xtc --view-mode coarse --smooth 5
cmd system.gro traj.xtc --smooth 7 --smoothres "protein"
Quick Start
Minimal launch:
cmd system.gro traj.xtc
cmd openmm_system.pdb traj.dcd
Protein-centric overview:
cmd system.gro traj.xtc --selection protein --view-mode cartoon
Membrane-system overview:
cmd system.gro traj.xtc --selection "protein or lipids or element K or element CL" --view-mode coarse
If you are using the test systems in this repository:
cmd test-trajs/traak/after90ns-k.pdb test-trajs/traak/traj_comp.xtc --selection protein --view-mode cartoon
cmd test-trajs/popc/em.gro test-trajs/popc/whole.xtc --view-mode coarse
cmd test-trajs/ga/reference-structure-2M-KCl.gro test-trajs/ga/04-pt7scaling-11pA.xtc --selection "resname K" --color-mode resid
For OpenMM-style trajectories:
cmd system.pdb trajectory.dcd
cmd system.pdb trajectory.dcd --selection protein --view-mode cartoon
cmd system.pdb trajectory.dcd --selection "protein or resname K" --color-mode resid
Controls
q: quith: toggle helpspace: play / pausenor right arrow: next framebor left arrow: previous framew/s: tilt cameraa/d: rotate cameraz/x: roll camerai/k: translate view up / downj/l: translate view left / right+/-: zoomr: reset camerao: toggle box overlay
The current zoom limit is 32x. Translation is screen-space panning, which is
useful when zoomed into a region after rotating the system.
Smoothing
--smooth Napplies centered trajectory smoothing overNframes.- By default smoothing targets proteins and lipid residues only.
--smoothres "selection"overrides which atoms are smoothed using the same MDAnalysis/VMD-style selection syntax as--selection.- Ions are intentionally excluded by default because they often move too far frame-to-frame for smoothing to look sensible.
Dynamic Water Overlay
--near-water 5shows water oxygens within5 Aof a target selection and updates that subset every frame.- The default target is
protein, and you can override it with--near-water-target "selection". - These waters are rendered as thin red glyphs so hydrated pockets and pore pathways are easier to spot without turning on bulk water.
Examples:
cmd system.gro traj.xtc --selection protein --near-water 5
cmd system.gro traj.xtc --view-mode cartoon --selection protein --near-water 4
cmd system.gro traj.xtc --near-water 5 --near-water-target "protein or lipids"
Representation Notes
pointsis still the best default for fast whole-system checks.traceworks best for proteins or nucleic-acid-heavy selections.coarseis intended for overview work: protein is shown as trace lines only, lipid residues try to anchor on headgroup atoms, and ions are colored by type in class mode following VMD-like expectations.cartoonis a protein-focused variant ofcoarsethat replaces protein dash traces with heuristic secondary-structure-aware ASCII: helix-like segments are thickened with@/o, sheet-like segments use directional arrows, and loops fall back to lighter=,|,/, and\backbone segments.--color-mode residis useful for tracking selected ions individually, for example--selection "resname K" --color-mode resid. Colors then cycle by residue id.- Box drawing currently supports orthorhombic unit cells only.
Examples:
cmd system.gro traj.xtc --view-mode points
cmd system.gro traj.xtc --view-mode trace --selection protein
cmd system.gro traj.xtc --view-mode coarse --selection "protein or lipids"
cmd system.gro traj.xtc --view-mode cartoon --selection protein
Selection notes
Selections are passed to MDAnalysis, which already supports the core concepts
needed here such as resname, name, resid, boolean operators, and ranges.
The viewer also normalizes atomname to name, and supports a built-in
lipids keyword that expands to a list of common lipid residue names.
Examples:
cmd system.gro traj.xtc --selection "protein"
cmd system.gro traj.xtc --selection "lipids"
cmd system.gro traj.xtc --selection "resname POPC or resname CHOL"
cmd system.gro traj.xtc --selection "protein or lipids"
cmd system.gro traj.xtc --selection "resid 10:50 and atomname CA"
cmd system.gro traj.xtc --selection "resname K"
cmd system.gro traj.xtc --selection "protein and around 5 resname K"
cmd system.gro traj.xtc --selection "resname POPC and name P"
Usage Recipes
Quick whole-system sanity check:
cmd system.gro traj.xtc --view-mode coarse
Check protein orientation in the membrane:
cmd system.gro traj.xtc --selection "protein or lipids" --view-mode coarse
Inspect secondary-structure-like protein shape:
cmd system.gro traj.xtc --selection protein --view-mode cartoon
Follow a specific ion type through a pore:
cmd system.gro traj.xtc --selection "resname K" --color-mode resid
cmd system.gro traj.xtc --selection "resname CL" --color-mode resid
Show only nearby hydration around the protein:
cmd system.gro traj.xtc --selection protein --near-water 5
Use smoothing for protein and lipid motions:
cmd system.gro traj.xtc --view-mode coarse --smooth 5
Smooth only the protein:
cmd system.gro traj.xtc --view-mode cartoon --smooth 7 --smoothres "protein"
Inspect a residue range or local region:
cmd system.gro traj.xtc --selection "resid 50:120"
cmd system.gro traj.xtc --selection "protein and around 6 resname LIG"
Roadmap
- improve playback performance for very large systems
- add residue / chain / segment centering shortcuts
- add small-selection ball-and-stick or stick-like modes
- support saved views and screenshots
- package for conda-based HPC installation
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file cmd_viewer-0.1.1.tar.gz.
File metadata
- Download URL: cmd_viewer-0.1.1.tar.gz
- Upload date:
- Size: 26.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f90f4a00e3f4a026d7d72490b23bfe0c3b64ffda7605473da78d90e7a0a848cd
|
|
| MD5 |
77126d1f32a0c105843428ffa28ba583
|
|
| BLAKE2b-256 |
e075d28036dbb900ebabb4be775f9a67453ea491857934f595f2d6a2fdd79362
|
Provenance
The following attestation bundles were made for cmd_viewer-0.1.1.tar.gz:
Publisher:
release.yml on Kopec-Lab/cmd-viewer
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
cmd_viewer-0.1.1.tar.gz -
Subject digest:
f90f4a00e3f4a026d7d72490b23bfe0c3b64ffda7605473da78d90e7a0a848cd - Sigstore transparency entry: 1507746699
- Sigstore integration time:
-
Permalink:
Kopec-Lab/cmd-viewer@a40edaa9d7926a48c9943d2538d425f575d447ac -
Branch / Tag:
refs/tags/v0.1.1 - Owner: https://github.com/Kopec-Lab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@a40edaa9d7926a48c9943d2538d425f575d447ac -
Trigger Event:
release
-
Statement type:
File details
Details for the file cmd_viewer-0.1.1-py3-none-any.whl.
File metadata
- Download URL: cmd_viewer-0.1.1-py3-none-any.whl
- Upload date:
- Size: 23.8 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ee5e0ece33364ce49ebdd9e941d580ea36a631a1bea6f850bc71691f5d020100
|
|
| MD5 |
f6dc43649ccb6306909d5edd38b3a8c9
|
|
| BLAKE2b-256 |
9999f861bcf05011d5d92e330600ee3965bb43cdd791e805510a432366faacab
|
Provenance
The following attestation bundles were made for cmd_viewer-0.1.1-py3-none-any.whl:
Publisher:
release.yml on Kopec-Lab/cmd-viewer
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
cmd_viewer-0.1.1-py3-none-any.whl -
Subject digest:
ee5e0ece33364ce49ebdd9e941d580ea36a631a1bea6f850bc71691f5d020100 - Sigstore transparency entry: 1507746788
- Sigstore integration time:
-
Permalink:
Kopec-Lab/cmd-viewer@a40edaa9d7926a48c9943d2538d425f575d447ac -
Branch / Tag:
refs/tags/v0.1.1 - Owner: https://github.com/Kopec-Lab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@a40edaa9d7926a48c9943d2538d425f575d447ac -
Trigger Event:
release
-
Statement type:







