DarkProfiler: Alignment and Classification of Peptides from Reference-Independent De Novo Peptide Sequencing Experiments.

Project description
DarkProfiler
DarkProfiler: Alignment and Classification of Peptides from Reference‑Independent De Novo Peptide Sequencing Experiments

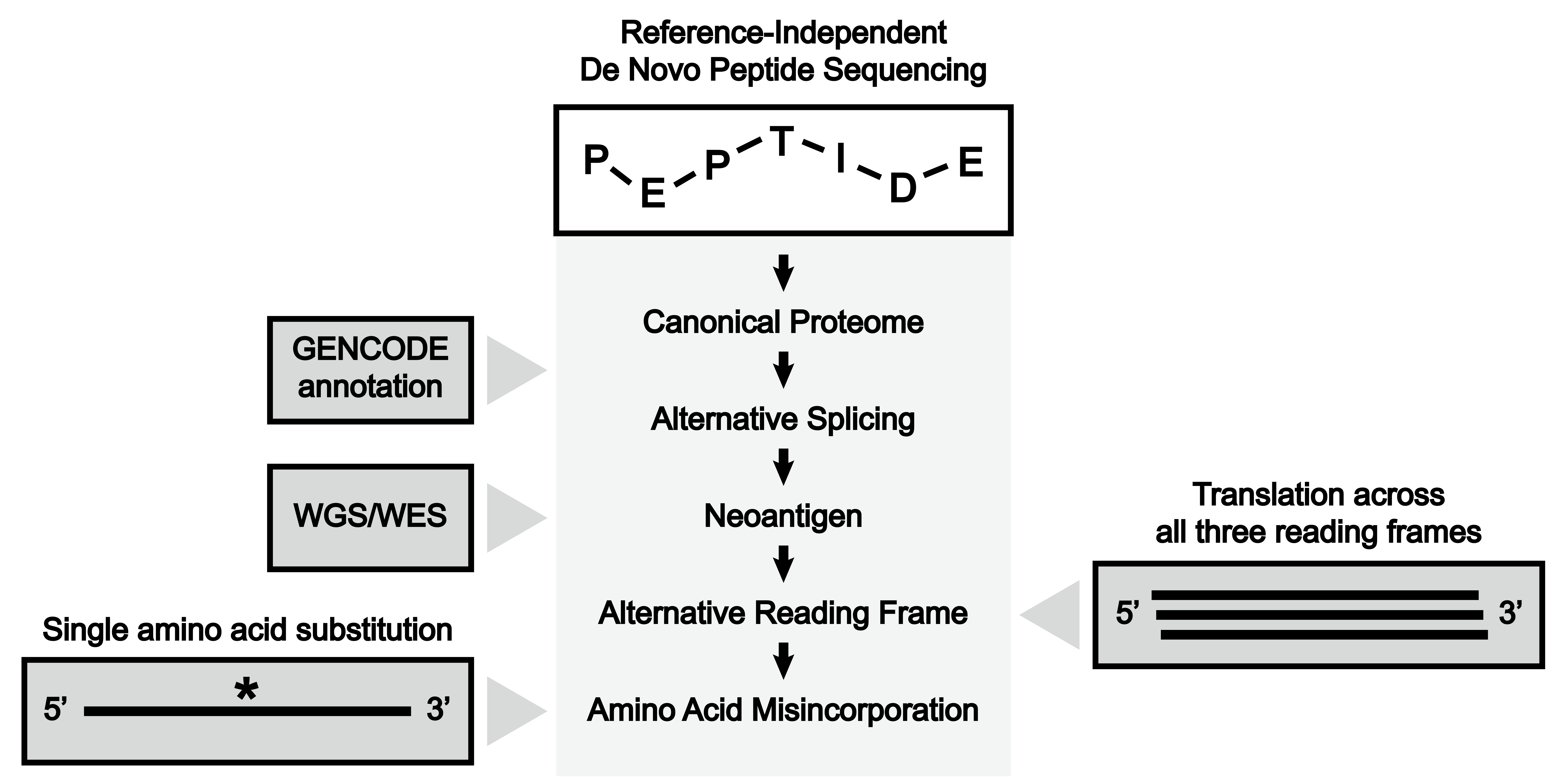
DarkProfiler takes peptide sequences (e.g., from reference-independent de novo peptide sequencing) and classifies them into distinct categories using reference genomes and optional sample‑specific SNVs:
- Canonical proteome
- Alternative splicing
- Neoantigens (SNV‑derived mutanome)
- Alternative reading frame peptides
- Amino acid misincorporations
- Unknown / unaligned
DarkProfiler is intended to be the post‑processing / annotation step after de novo peptide calling or customized peptide discovery in proteomics or immunopeptidomics experiments.
Supported reference assemblies:
- Human:
hg19(GENCODE release 19),hg38(GENCODE release 37) - Mouse:
mm10(GENCODE release M19),mm39(GENCODE release M37)
The same logic is available both as a command‑line tool and as a Python API.
Table of contents
- Installation
- Reference genome data
- Input data
- Command‑line usage
- Python API
- Classification pipeline details
- Outputs
- Database reuse and performance tips
- Troubleshooting
- License
- Citation
Installation
Requirements
- Python: 3.7+ (tested on modern CPython versions)
- Operating systems: Linux, macOS, and other UNIX‑like systems should work. Windows with WSL is recommended.
- Python dependencies (installed automatically via pip/conda):
- Biopython (FASTA parsing and sequence utilities)
- matplotlib (for
pieChart.pdf) - Standard library modules only otherwise
You also need sufficient disk space to store:
- A reference genome bundle per assembly (hundreds of MB)
- The database directory (translated proteomes) per output folder
- The final classification FASTA files and plots
Install with pip (PyPI)
pip install darkprofiler
This installs:
- The Python package
darkprofiler - The command‑line entry point
darkprofiler
You should then be able to run:
darkprofiler --help
Install with conda (bioconda)
conda install bioconda::darkprofiler
This will install DarkProfiler together with all dependencies into the active conda environment.
Reference genome data
Supported references
DarkProfiler currently supports human and mouse reference assemblies that are aligned to GENCODE releases:
hg19 (GENCODE release 19)
hg38 (GENCODE release 37)
mm10 (GENCODE release M19)
mm39 (GENCODE release M37)
The reference is always specified by one of the lower‑case strings:
hg19hg38mm10mm39
Internally the reference is normalized to lower case, so HG38 and hg38 are treated the same in the Python API, but the CLI restricts choices to the canonical lower‑case names.
What gets downloaded
Reference data are distributed as versioned ZIP bundles hosted online. You do not need to download or unpack them manually. Use:
darkprofiler download hg38
This will:
-
Check that the requested reference is supported.
-
Download a file named like
darkprofiler_hg38.zipto the installed package directory underdarkprofiler/genome/. -
Extract the contents to:
<python-site-packages>/darkprofiler/genome/hg38/
-
Print progress messages such as:
[darkprofiler] Downloading ... [darkprofiler] Extracting to ... [darkprofiler] Finished. Reference 'hg38' is now available.
The extracted directory contains at least the following files (names may include version tags):
transcriptome.<reference>.fa– all reference transcripts (FASTA)transcriptome.<reference>.cds.bed– CDS segments per transcriptknownCanonical.<reference>.list– list of canonical transcript IDsgencode.<reference>.gff– GENCODE annotation (GFF/GTF‑like)exome.<reference>.bed– exome intervals used to filter SNVs
These files are used internally by the pipeline; you normally don’t need to interact with them directly.
Note: If the
downloadstep has not been run for a given reference,darkprofiler runwill fail with an error such as “Could not find file ... in genome root”.
Input data
Peptide FASTA
The primary input is a FASTA file containing peptide sequences to classify:
>peptide_1
LLLLGIGGTFK
>peptide_2
EAVAEQAALR
...
Requirements and recommendations:
- Each record is interpreted as a peptide (amino‑acid sequence).
- FASTA IDs are kept as‑is and propagated to the output files.
- Sequences are upper‑cased internally; non‑standard characters are not specially treated.
- Empty sequences are silently ignored.
- There is no hard limit on peptide length, but short peptides may match many locations and very long peptides may be rare.
The same peptide ID will appear in at most one output FASTA file, corresponding to the first category that matches in the pipeline (canonical → alternative splicing → neoantigen → alternative reading frame → misincorporation → unknown).
VCF with SNVs (optional)
To classify neoantigens (peptides derived from sample‑specific single nucleotide variants), you can provide a VCF file via --vcf-path / vcf_path:
- Accepts plain or gzipped VCF:
*.vcfor*.vcf.gz. - Only SNVs (single‑base reference and single‑base alternate) are used.
- Multi‑allelic entries are expanded and processed per ALT allele.
- Non‑SNV variants (indels, MNVs, etc.) are ignored.
- Coordinates are matched to the reference via chromosome names that are normalized to strip the
chrprefix (chr1→1).
DarkProfiler additionally filters SNVs to the coding exome using the exome.<reference>.bed file if present:
- Only SNVs whose positions overlap the exome intervals are retained.
- If no exome BED is available, all SNVs are accepted.
If vcf_path is omitted or points to a non‑existing file:
- The SNV list is empty.
- The “mutanome” and “neoantigen” step still runs but reduces to the canonical proteome (no sample‑specific variation).
- Classification still works; you simply will not obtain any neoantigen‑specific hits beyond what is already canonical.
Precomputed database directory (optional)
By default, each darkprofiler run invocation builds a database in:
<output_dir>/database/
The database contains translated and derived proteomes as FASTA files:
canonicalProteome.faalternativeSplicing.famutanome.famutatedCanonicalTranscriptome.famutatedAlternativeTranslatome.famutatedAlternativeORFeome.fa
If you run DarkProfiler repeatedly with the same reference and SNV set, you can re‑use a prebuilt database to avoid recomputation by passing --database-path / database_path:
darkprofiler run hg38 peptides.fa out --database-path prebuilt_db/
The directory is accepted only if all required files are present. Otherwise:
- DarkProfiler prints a warning that the directory is missing files or is invalid.
- The directory is ignored.
- A new database is built from scratch under
<output_dir>/database.
Re‑using databases is optional, but can substantially speed up repeated analyses on the same genotype.
Command‑line usage
The installed CLI is called darkprofiler.
Run darkprofiler --help to see the top‑level usage:
usage: darkprofiler [-h] {download,run} ...
DarkProfiler: classify peptides into canonical, alternative, mutant,
and dark proteome categories.
Two subcommands are available:
darkprofiler download– download reference genome bundles.darkprofiler run– run the classification pipeline.
download subcommand
darkprofiler download hg38
Positional arguments
-
reference(choices:hg19,hg38,mm10,mm39)Reference assembly version to download. The download is performed once per environment; re‑running will simply re‑use the existing files.
run subcommand
darkprofiler run hg38 peptides.fa output_dir --vcf-path sample.vcf.gz --database-path /path/to/database --num-threads 8
Positional arguments
-
reference(choices:hg19,hg38,mm10,mm39)Reference assembly version to use. The corresponding reference bundle must have been downloaded beforehand with
darkprofiler download. -
peptide_fastaPath to peptide FASTA file (input peptides to classify).
-
output_dirOutput directory. Will be created if it does not exist. All category FASTAs and summary files are written here.
Optional arguments
-
--vcf-path FILEOptional path to a VCF or VCF.GZ file with SNVs. When provided and valid, SNVs are mapped through the transcriptome to construct a mutated canonical transcriptome and mutanome. Peptides mapping uniquely to the mutanome become neoantigens.
-
--database-path DIROptional path to an existing database directory containing:
canonicalProteome.faalternativeSplicing.famutanome.famutatedCanonicalTranscriptome.famutatedAlternativeTranslatome.famutatedAlternativeORFeome.fa
If the directory is valid and complete, it is reused directly, skipping database construction. If any required file is missing or the path is invalid, a warning is printed and DarkProfiler rebuilds the database in
<output_dir>/database. -
--num-threads N(default:1)Number of threads for the amino acid misincorporation search. Only this step is parallelised. Values ≤ 1 run single‑threaded.
Progress and logging
The pipeline prints a 10‑step progress bar to stderr, for example:
[##########------------------------------] 3/10 - Build canonical / non-canonical transcript sets
Within some steps (e.g. canonical classification, alternative splicing, mutanome, etc.), additional per‑100‑peptide progress bars are printed to stderr.
Normal output files are written to output_dir and do not interleave with the log messages.
Examples
Minimal run (no SNVs, new database per run):
darkprofiler download hg38
darkprofiler run hg38 peptides.fa results/
Run with SNVs:
darkprofiler download hg38
darkprofiler run hg38 peptides.fa results/ --vcf-path tumor_sample.vcf.gz --num-threads 4
Re‑use a precomputed database (same reference and SNVs):
# First run builds the database under results/database
darkprofiler download mm39
darkprofiler run mm39 peptides.fa results/ --vcf-path sample.vcf.gz
# Subsequent runs can reuse that database
darkprofiler run mm39 other_peptides.fa new_results/ --database-path results/database --vcf-path sample.vcf.gz
Python API
DarkProfiler exposes the same functionality via a Python function.
Function reference
from darkprofiler.run import classify_peptides
classify_peptides(
reference="hg38",
peptide_fasta="peptides.fa",
output_dir="output",
vcf_path=None,
database_path=None,
num_threads=4,
)
Parameters
-
reference: strReference assembly to use. One of:
"hg19","hg38","mm10","mm39"(case‑insensitive). Any other value raises aValueError. -
peptide_fasta: strPath to the peptide FASTA file to classify.
-
output_dir: strOutput directory. Created if missing. Classification FASTAs, the database (unless reusing one), and summary files are written here.
-
vcf_path: Optional[str](default:None)Path to a VCF or VCF.GZ file containing SNVs. If
Noneor the file does not exist, the SNV list is empty and the mutanome step reduces to the canonical proteome (i.e. no sample‑specific neoantigens). -
database_path: Optional[str](default:None)Path to an existing database directory. If valid and complete, the directory is reused and database construction is skipped. Otherwise, a new database directory is created under
output_dirand filled. -
num_threads: int(default:1)Number of threads used only in the amino acid misincorporation search (a Hamming distance ≤ 1 search against alternative ORFs). Values ≤ 1 run single‑threaded.
The function prints progress to stderr and returns None. All results are materialized as files on disk.
Python examples
Basic usage from a script:
from darkprofiler.run import classify_peptides
classify_peptides(
reference="hg38",
peptide_fasta="peptides.fa",
output_dir="results",
vcf_path="sample.vcf.gz",
database_path=None,
num_threads=8,
)
Reusing a database directory from Python:
from darkprofiler.run import classify_peptides
# Suppose "db" already contains the six required FASTA files
classify_peptides(
reference="mm10",
peptide_fasta="new_peptides.fa",
output_dir="run2",
vcf_path="sample.vcf.gz",
database_path="db",
num_threads=4,
)
Running programmatically without installing the CLI (e.g. in a notebook) is also supported as long as the reference genome has already been downloaded via the darkprofiler download command in your environment.
Classification pipeline details
Overview of steps
The internal pipeline consists of the following conceptual steps (as printed in the progress bar):
-
Filter VCF to exome
Load the exome BED, parse the VCF, normalize chromosome names, keep SNVs that fall in exonic intervals. -
Setup and load transcriptome/CDS/knownCanonical
Load the transcriptome FASTA, CDS BED, and the list of canonical transcript IDs for the chosen reference. -
Build canonical / non‑canonical transcript sets
Split transcript IDs into canonical vs non‑canonical groups using the canonical list. -
Generate canonical proteome and classify canonical peptides
Translate CDS for canonical transcripts into the canonical proteome; classify peptides that match exactly. -
Generate alternative splicing proteome and classify peptides
Translate CDS for non‑canonical transcripts (e.g. splice isoforms); classify peptides that match exactly. -
Apply SNVs, generate mutanome and classify neoantigens
Apply exonic SNVs to canonical transcripts, translate CDS, and classify peptides that match the resulting mutanome proteome but not the canonical ones. -
Generate alternative ORFs and classify peptides
Translate all three reading frames of the mutated canonical transcriptome; classify peptides that match these alternative reading frames. -
Identify amino acid misincorporations
Search for peptide sequences that differ from any alternative ORF by at most one amino acid (Hamming distance ≤ 1). These are classified as amino acid misincorporations. -
Write unaligned peptides and pie chart
Any peptides still unclassified are written tounknown.fa. Category counts are summarized intopieChart.tsvand visualized as a pie chart PDF. -
Finalize
Cleanup and final progress message.
Category definitions
Below, “remaining peptides” refers to the set of peptides that have not yet been classified in previous steps.
1. Canonical proteome (canonicalProteome.fa)
- Proteins derived by translating CDS regions of canonical transcripts only.
- Peptides that match exactly (substring match) anywhere within any canonical protein are assigned to the canonical proteome category.
- Output FASTA:
canonicalProteome.fainoutput_dir:- FASTA IDs: original peptide ID followed by
|and the matched canonical transcript ID.
- FASTA IDs: original peptide ID followed by
2. Alternative splicing (alternativeSplicing.fa)
- Proteins derived by translating CDS regions of non‑canonical transcripts (e.g. alternative splice forms).
- Remaining peptides that match exactly any of these proteins are classified as alternative splicing hits.
- Output FASTA:
alternativeSplicing.fa.
3. Neoantigens (neoantigen.fa)
- First, SNVs are mapped to canonical transcripts using GENCODE exon annotations and strand information.
- For each canonical transcript, the exonic sequence is reconstructed, SNVs are applied in transcript coordinates, and CDS is translated to form a mutated canonical proteome (mutanome).
- Remaining peptides that match exactly any protein in the mutanome are classified as neoantigens:
- These represent peptides that can arise only due to sample‑specific SNVs (or that coincide with canonical regions when no SNVs are present).
- Output FASTA:
neoantigen.fa.
Peptides are matched by simple substring search; no alignment or scoring is performed at this stage.
4. Alternative reading frame peptides (alternativeReadingFrame.fa)
- For each mutated canonical transcript, DarkProfiler translates all three reading frames (frame 0, 1, 2) over the full transcript sequence, not just CDS.
- These frame translations are written into the alternative ORF proteome (
mutatedAlternativeTranslatome.faandmutatedAlternativeORFeome.fain the database). - Remaining peptides that match exactly any of these frame‑translated proteins are classified as alternative reading frame peptides.
- Output FASTA:
alternativeReadingFrame.fa.
This captures peptides that may arise from alternative translation initiation, frameshifts, or unannotated ORFs.
5. Amino acid misincorporations (aminoAcidMisincorporation.fa)
- For peptides still unclassified, DarkProfiler tests whether they differ from any alternative ORF peptide by at most 1 amino acid using a Hamming‑distance‑based approach:
- For each peptide, all sequences with Hamming distance ≤ 1 (including the original) are generated.
- These variants are searched as substrings within each alternative ORF protein sequence.
- If any such variant occurs in an alternative ORF, the peptide is classified as an amino acid misincorporation.
- Output FASTA:
aminoAcidMisincorporation.fa.
This category is intended to capture likely translation or sequencing errors where a peptide is nearly canonical / alternative but differs by one residue.
6. Unknown (unknown.fa)
Peptides that do not fall into any of the above categories are written unmodified to:
unknown.fa
These may represent:
- Completely novel proteomic events
- Peptides arising from structural variants or indels
- Database or reference limitations
- False positives from upstream de novo sequencing
Outputs
All outputs live in the specified output_dir and are overwritten if you re‑run the pipeline with the same directory.
FASTA category files
Each category is represented by a separate FASTA file in output_dir:
canonicalProteome.faalternativeSplicing.faneoantigen.faalternativeReadingFrame.faaminoAcidMisincorporation.faunknown.fa
Each header line contains the original peptide ID, and, when available, the reference source identifier, for example:
>pep0001 | ENST00000335137
SEQUENCEHERE
This makes it easy to join back to upstream metadata tables or downstream visualization tools.
pieChart.tsv
A tab‑separated summary file with one line per category:
Category Count
canonical 123
alternativeSplicing 45
neoantigen 7
alternativeReadingFrame 32
aminoAcidMisincorporation 10
unknown 83
The categories follow this fixed order:
canonicalalternativeSplicingneoantigenalternativeReadingFrameaminoAcidMisincorporationunknown
You can import this file into R, Python, or a spreadsheet program to generate additional plots or statistics.
pieChart.pdf
A publication‑quality pie chart illustrating the fraction of peptides in each category is saved as:
pieChart.pdf
Key details:
-
Generated via
matplotlibwith high resolution (dpi=1200). -
Fixed color scheme (hex colors):
- canonical proteome –
#263b81 - alternative splicing –
#0578a6 - neoantigen –
#64cdf6 - alternative reading frame –
#d71f26 - amino acid misincorporation –
#f493a9 - unknown –
#e5e5e5
- canonical proteome –
-
A legend shows human‑readable category names: “canonical proteome”, “alternative splicing”, “neoantigen”, etc.
-
Categories with count 0 are omitted from the pie but still shown in the legend.
If all counts are zero (e.g. an empty input FASTA), the pie chart is skipped.
Database reuse and performance tips
-
Reusing the database
For repeated analyses on the same reference and SNV set, use--database-pathto reuse a previously built database. This avoids re‑translating transcriptomes and applying SNVs. -
Multi‑threading
The most computationally intensive step is the amino acid misincorporation search, which scales with the number of peptides and the size of the alternative ORF proteome. Use--num-threadsto parallelize this step on multi‑core machines. -
Peptide batching
If you have a very large peptide set, you can split your FASTA into chunks and process them in separate runs, then combine the output FASTAs downstream. -
Disk space
The database directory may contain multiple large FASTA files. If disk space is a concern, you can delete or compress database directories once you are done, and rebuild them later if needed.
Troubleshooting
“Unsupported reference 'XXX'”
- The reference must be one of
hg19,hg38,mm10,mm39. Check for typos or capitalization. The CLI enforces the allowed values.
“Could not find file ... in genome root” or missing GENCODE/GFF/CDS files
- Make sure you have run
darkprofiler download <reference>for the same Python environment where you are running the pipeline. - Verify that you are using the correct reference name.
No neoantigen hits
- Ensure that:
--vcf-pathpoints to the correct sample VCF.- The VCF contains SNVs overlapping the exome of the chosen reference.
- Remember that only SNVs are currently applied; indels and complex variants are ignored.
Database path ignored with a warning
- If
--database-pathis provided but any of the required files are missing, DarkProfiler prints a warning and rebuilds the database inoutput_dir/database. Make sure the directory is complete and originates from a previous successful run.
Large runtime or memory usage
- Increase
--num-threadsto speed up misincorporation search on multi‑core machines. - Reduce the input peptide set (e.g. filter for high‑confidence de novo calls).
- Reuse databases where possible to skip the expensive SNV application and frame translation steps.
If you encounter issues that are not addressed here, consider inspecting the STDERR logs for warnings (e.g. reference base mismatches when applying SNVs) and double‑checking that all inputs are aligned to the same reference assembly.
License
DarkProfiler is released under the MIT License.
MIT License
Copyright (c) 2025
Citation
If you use DarkProfiler in a scientific publication, please cite it as:
DarkProfiler: alignment and classification of peptides from reference‑independent de novo peptide sequencing experiments. 2025.
(Updated citation information will be provided once an associated preprint or manuscript is available.)
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file darkprofiler-0.1.2.tar.gz.
File metadata
- Download URL: darkprofiler-0.1.2.tar.gz
- Upload date:
- Size: 30.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.4.2 requests/2.28.2 setuptools/45.2.0 requests-toolbelt/0.8.0 tqdm/4.67.1 CPython/3.8.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8a52691fa7a269253e14289004bc8b711d152e5316cf1fb0e0adccf8c52f5ac8
|
|
| MD5 |
a6e24216aa1f0180b99875ccb8e66f3e
|
|
| BLAKE2b-256 |
f7feb51c9514f5909e478b60afd0fb49b3b199c2d792eed663f8ee14c4007dd2
|
File details
Details for the file darkprofiler-0.1.2-py3-none-any.whl.
File metadata
- Download URL: darkprofiler-0.1.2-py3-none-any.whl
- Upload date:
- Size: 22.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.4.2 requests/2.28.2 setuptools/45.2.0 requests-toolbelt/0.8.0 tqdm/4.67.1 CPython/3.8.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1cd92488ead9f1f5f27c74dd229a2c230ae3ff6d2b37c3415c7f6aec729491e1
|
|
| MD5 |
0a42d5b53c73ce359b842fa80ab72c11
|
|
| BLAKE2b-256 |
e08ebb43813671cb6ed5611566b009cad1e201ae22eb450d3236fdd8ad05ad37
|







