DarkProfiler: Alignment and Classification of Peptides from Reference-Independent De Novo Peptide Sequencing Experiments.

Project description
DarkProfiler
DarkProfiler: Alignment and Classification of Peptides from Reference‑Independent De Novo Peptide Sequencing Experiments

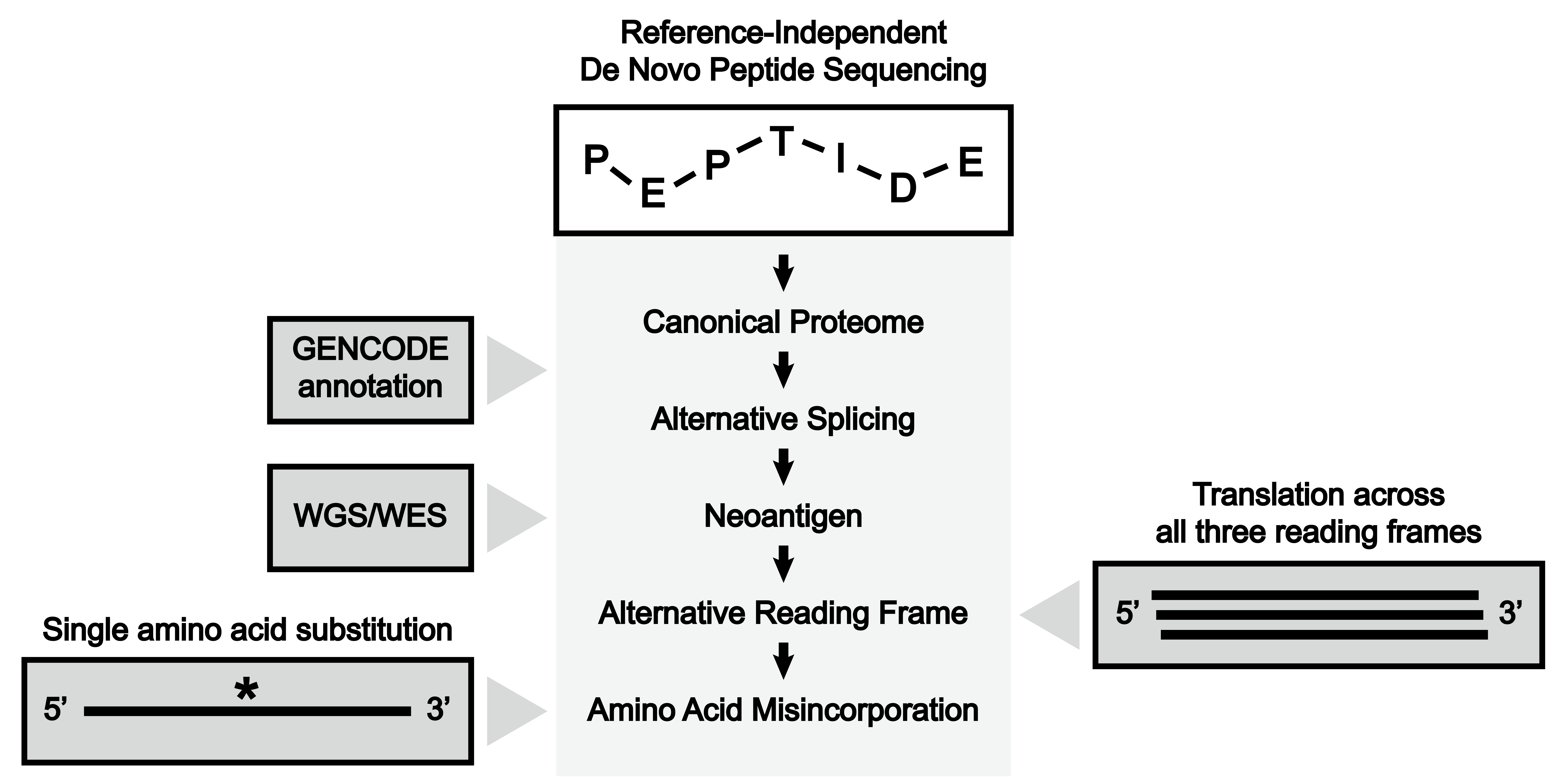
DarkProfiler takes peptide sequences (e.g., from reference‑independent de novo peptide sequencing) and classifies them into distinct categories using reference genomes and optional sample‑specific SNVs:
- Canonical proteome
- Alternative splicing
- Neoantigens (SNV‑derived mutanome)
- Alternative reading frame peptides
- Unknown / unaligned
DarkProfiler is intended to be the post‑processing / annotation step after de novo peptide calling or customized peptide discovery in proteomics or immunopeptidomics experiments.
Supported reference assemblies:
- Human:
hg19(GENCODE release 19),hg38(GENCODE release 37) - Mouse:
mm10(GENCODE release M19),mm39(GENCODE release M37)
The same logic is available both as a command‑line tool and as a Python API.
Table of contents
- Installation
- Reference genome data
- Input data
- Command‑line usage
- Python API
- Classification pipeline details
- Outputs
- Database reuse and performance tips
- Troubleshooting
- License
- Citation
Installation
Requirements
- Python: 3.7+ (tested on modern CPython versions)
- Operating systems: Linux, macOS, and other UNIX‑like systems should work. Windows with WSL is recommended.
- Python dependencies (installed automatically via pip/conda):
- Biopython (FASTA parsing and sequence utilities)
- matplotlib (for
pieChart.pdf) - Standard library modules only otherwise
You also need sufficient disk space to store:
- A reference genome bundle per assembly (hundreds of MB)
- The database directory (translated proteomes + fast indices) per output folder
- The final classification FASTA files and plots
Install with pip (PyPI)
pip install darkprofiler
This installs:
- The Python package
darkprofiler - The command‑line entry point
darkprofiler
You should then be able to run:
darkprofiler --help
Install with conda (bioconda)
conda install bioconda::darkprofiler
This will install DarkProfiler together with all dependencies into the active conda environment.
Reference genome data
Supported references
DarkProfiler currently supports human and mouse reference assemblies that are aligned to GENCODE releases:
hg19 (GENCODE release 19)
hg38 (GENCODE release 37)
mm10 (GENCODE release M19)
mm39 (GENCODE release M37)
The reference is always specified by one of the lower‑case strings:
hg19hg38mm10mm39
Internally the reference is normalized to lower case, so HG38 and hg38 are treated the same in the Python API, but the CLI restricts choices to the canonical lower‑case names.
What gets downloaded
Reference data are distributed as versioned ZIP bundles hosted online. You do not need to download or unpack them manually. Use:
darkprofiler download hg38
This will:
-
Check that the requested reference is supported.
-
Download a file named like
darkprofiler_hg38.zipto the installed package directory underdarkprofiler/genome/. -
Extract the contents to:
<python-site-packages>/darkprofiler/genome/hg38/
-
Print progress messages such as:
[darkprofiler] Downloading ... [darkprofiler] Extracting to ... [darkprofiler] Finished. Reference 'hg38' is now available.
The extracted directory contains at least the following files (names may include version tags):
transcriptome.<reference>.fa– all reference transcripts (FASTA)transcriptome.<reference>.cds.bed– CDS segments per transcriptknownCanonical.<reference>.list– list of canonical transcript IDsgencode.<reference>.gff– GENCODE annotation (GFF/GTF‑like)exome.<reference>.bed– exome intervals used to filter SNVs
These files are used internally by the pipeline; you normally don’t need to interact with them directly.
Note: If the
downloadstep has not been run for a given reference,darkprofiler runwill fail with an error such as “Could not find file ... in genome root”.
Input data
Peptide FASTA
The primary input is a FASTA file containing peptide sequences to classify:
>peptide_1
LLLLGIGGTFK
>peptide_2
EAVAEQAALR
...
Requirements and recommendations:
- Each record is interpreted as a peptide (amino‑acid sequence).
- FASTA IDs are kept as‑is and propagated to the output files.
- Sequences are upper‑cased internally; non‑standard characters are not specially treated.
- Empty sequences are silently ignored.
- There is no hard limit on peptide length, but short peptides may match many locations and very long peptides may be rare.
A peptide sequence is assigned to at most one output category within a given hamming distance, corresponding to the first category that matches in the pipeline (canonical → alternative splicing → neoantigen → alternative reading frame → unknown).
VCF with SNVs (optional)
To classify neoantigens (peptides derived from sample‑specific single nucleotide variants), you can provide a VCF file via --vcf-path / vcf_path:
- Accepts plain or gzipped VCF:
*.vcfor*.vcf.gz. - Only SNVs (single‑base reference and single‑base alternate) are used.
- Multi‑allelic entries are expanded and processed per ALT allele.
- Non‑SNV variants (indels, MNVs, etc.) are ignored.
- Coordinates are matched to the reference via chromosome names that are normalized to strip the
chrprefix (chr1→1).
DarkProfiler additionally filters SNVs to the coding exome using the exome.<reference>.bed file if present:
- Only SNVs whose positions overlap the exome intervals are retained.
- If no exome BED is available, all SNVs are accepted.
If vcf_path is omitted or points to a non‑existing file:
- The SNV list is empty.
- The mutanome and neoantigen steps still run, but represent the unmodified reference sequence.
Precomputed database directory (optional)
By default, each darkprofiler run invocation builds a database in:
<output_dir>/database/
The database contains translated and derived proteomes as FASTA files:
canonicalProteome.faalternativeSplicing.famutanome.famutatedCanonicalTranscriptome.famutatedAlternativeTranslatome.fa
DarkProfiler also creates persistent fast indices under the same database directory to accelerate peptide search with Hamming distance: for example:
canonicalProteome.idx/alternativeSplicing.idx/mutanome.idx/mutatedAlternativeORFeome.idx/
If you run DarkProfiler repeatedly with the same reference and SNV set, you can re‑use a prebuilt database to avoid recomputation by passing --database-path / database_path:
darkprofiler run hg38 peptides.fa out --database-path prebuilt_db/
The directory is accepted only if all required files are present. Otherwise:
- DarkProfiler prints a warning that the directory is missing files or is invalid.
- The directory is ignored.
- A new database is built from scratch under
<output_dir>/database.
Command‑line usage
The installed CLI is called darkprofiler.
Run darkprofiler --help to see the top‑level usage:
usage: darkprofiler [-h] {download,run} ...
Two subcommands are available:
darkprofiler download– download reference genome bundles.darkprofiler run– run the classification pipeline.
download subcommand
darkprofiler download hg38
run subcommand
darkprofiler run hg38 peptides.fa output_dir \
--vcf-path sample.vcf.gz \
--database-path /path/to/database \
--num-threads 8 \
--hamming 2
Optional arguments
-
--vcf-path FILEOptional path to a VCF or VCF.GZ file with SNVs.
-
--database-path DIROptional path to an existing database directory containing the required FASTA files listed above.
-
--num-threads N(default:1)Number of worker threads used during peptide search / verification.
-
-k, --hamming {0,1,2}(default:0)Maximum Hamming distance allowed for peptide matching.
0performs exact matches only;1and2allow up to one or two amino‑acid substitutions.
Python API
from darkprofiler.run import classify_peptides
classify_peptides(
reference="hg38",
peptide_fasta="peptides.fa",
output_dir="output",
vcf_path=None,
database_path=None,
num_threads=4,
hamming_distance=0,
)
Classification pipeline details
Overview of steps
- Filter VCF to exome
- Load transcriptome, CDS annotations, canonical transcript list
- Build canonical / non‑canonical transcript sets
- Build canonical proteome (CDS must start with
ATG) and classify peptides - Build alternative splicing proteome (CDS must start with
ATG) and classify peptides - Apply SNVs, build mutanome (CDS must start with
ATG) and classify peptides - Build alternative ORFs (3 frames) and classify peptides
- Write unaligned peptides and summary plots
- Finalize
Category definitions
-
CDS translation filter (
ATG)
For CDS‑based proteomes (canonical proteome, alternative splicing, mutanome), CDS translations are included only when the CDS begins withATG. This reduces false positives from incomplete or mis‑annotated CDS records. -
ORF region labels
For alternative ORF hits, DarkProfiler labels the peptide start as:uORF(upstream of CDS start)intORF(out-of-frame peptdies from inside annotated CDS span)dORF(downstream of CDS end)lncRNA(no CDS annotation)
Outputs
All outputs live in the specified output_dir.
FASTA category files
Each category is represented by a separate FASTA file in output_dir:
canonicalProteome.faalternativeSplicing.faneoantigen.faalternativeReadingFrame.faunknown.fa
For classification FASTAs (all except unknown.fa), each record uses:
> referencePeptide | TranscriptID | nucleotide coordinate on transcript | uORF/intORF/dORF/lncRNA/CDS
queryPeptide
- referencePeptide: matched reference peptide sequence (substring from the reference proteome/ORF; same length as the query)
- TranscriptID: transcript identifier (for alternative ORFs, this is the underlying transcript)
- nucleotide coordinate on transcript: 1‑based transcript coordinate of the peptide start codon (frame‑aware for alternative ORFs)
- uORF/intORF/dORF/lncRNA/CDS:
CDSfor canonical proteome / alternative splicing / neoantigen hitsuORF,intORF,dORF,lncRNAfor alternative ORF hits
Example:
> GILGFVFTL | ENST00000335137.4 | 1234 | CDS
GILGFVFTL
unknown.fa uses the original peptide IDs and sequences without additional fields.
pieChart.tsv
A tab‑separated summary file with one line per category:
Category Count
canonical 123
alternativeSplicing 45
neoantigen 7
alternativeReadingFrame 32
unknown 83
pieChart.pdf
A pie chart illustrating the fraction of peptides in each category is saved as pieChart.pdf.
Database reuse and performance tips
-
Reuse databases
Use--database-pathto reuse a database directory containing the required FASTA files. -
Persistent fast indices
DarkProfiler builds on‑disk indices (*.idx/) for fast peptide lookup with Hamming distance ≤ 2 using a pigeonhole (seed‑and‑verify) strategy. When an index directory exists, it is reused automatically. -
Multi‑threading
Increase--num-threadsto speed up peptide search / verification on multi‑core machines.
Troubleshooting
Unsupported reference
- The reference must be one of
hg19,hg38,mm10,mm39.
Missing genome files
- Run
darkprofiler download <reference>in the same environment.
Large runtime
- Increase
--num-threads. - Use
-k/--hamming 0for exact matching only when appropriate. - Reuse databases and indices between runs.
License
DarkProfiler is released under the MIT License.
Citation
If you use DarkProfiler in a scientific publication, please cite it as:
(Updated citation information will be provided once an associated preprint or manuscript is available.)
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file darkprofiler-0.2.4.tar.gz.
File metadata
- Download URL: darkprofiler-0.2.4.tar.gz
- Upload date:
- Size: 26.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.4.2 requests/2.28.2 setuptools/45.2.0 requests-toolbelt/0.8.0 tqdm/4.67.1 CPython/3.8.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
72755950c343d58deb7cdedf03ab14dab500bd318469adc8dcce33f8e9570fa2
|
|
| MD5 |
074855aac54a505bd67f5064abf5fffc
|
|
| BLAKE2b-256 |
9406d60bfa6314f6c4aa6615c48b05fcb7cfcf7572dfbf43a75ea1b281d68538
|
File details
Details for the file darkprofiler-0.2.4-py3-none-any.whl.
File metadata
- Download URL: darkprofiler-0.2.4-py3-none-any.whl
- Upload date:
- Size: 21.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.4.2 requests/2.28.2 setuptools/45.2.0 requests-toolbelt/0.8.0 tqdm/4.67.1 CPython/3.8.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3f918810b7121f3aecc3297f8da1fe11eb1d06fcdac561a505c6a1ef90a43a24
|
|
| MD5 |
e99f252d182c9ebc3511eb009fdb8365
|
|
| BLAKE2b-256 |
c2e4fa96ceb41ebb12b9d963636163126f003c71bd8ee2cd5f13553030193820
|







