gtracks
Plot genome track data from bigWig and bedGraph files. Powered by pyGenomeTracks.
Installation
pip install gtracks
or
pip install --user gtracks
In conda
conda create -n gtracks -c conda-forge -c bioconda deeptools seaborn pybedtools gff2bed
conda activate gtracks
mamba install -c conda-forge -c bioconda pygenometracks
pip install gtracks
Examples
An example bigwig file with ATAC-seq data from the insulin region is included. You can generate a test plot like this:
gtracks INS-IGF2 test.png
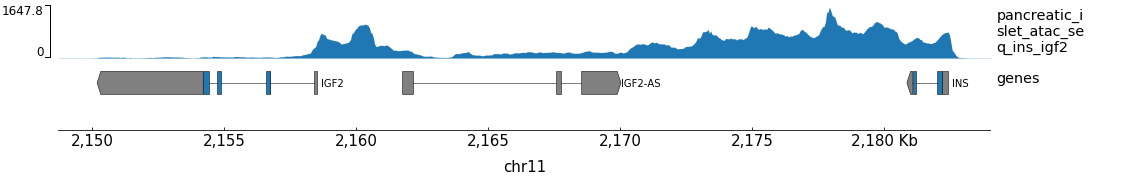
You can plot your own tracks over other genomic regions by providing more positional arguments: a region or gene name and paths to one or more bigWig files. The file type of the plot will be determined by the output file extension.
gtracks chr11:2150341-2182439 track1.bw track2.bw output.pdf
gtracks INS track1.bw track2.bw output.svg
Modifying the gene annotations track
GRCh37/hg19 gene annotations are used by default, but you can plot GRCh38/hg38
genes by adding --genes GRCh38 or --genes hg38. You can use your own gene
annotations file (BED or BED12 format) by providing
--genes <path/to/genes.bed.gz>.
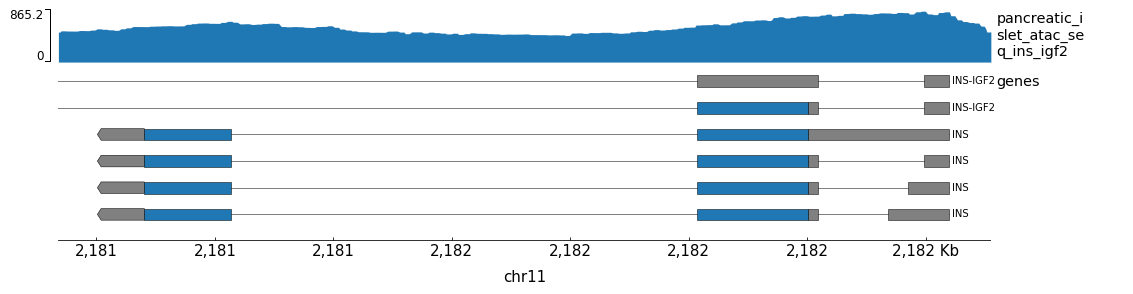
You may want to add more rows to the genes track. You can do this using
the --genes-height and --gene-rows options.
gtracks INS test-genes.png --genes-height 6 --gene-rows 6
Coordinate parsing
By default, gtracks parses input coordinates according to the following regular expression:
([Cc]hr)?[0-9XYZWM]+:[0-9]+-[0-9]+$
Inputs not matching this expression are interpreted as gene names. This can cause errors
if e.g. your contig names do not match the format. To change how coordinates are parsed,
you can supply an alternative regular expression using the --coord-regex option. For example, the following expression
allows arbitrary contig names:
--coord-regex '[\s\S]+:[0-9]+-[0-9]+$'
Changing the color palette
You can change the color palette for bigWig tracks using the --color-palette option.
gtracks INS track1.bw track2.bw track3.bw output.pdf --color-palette "#color1" "#color2" "#color3"
Setting y-axis height
By default, tracks have different y-axis heights depending on signal height.
You can set a uniform y-axis height for all tracks using the --max option.
gtracks INS track1.bw track2.bw track3.bw output.pdf --max 400
For more command-line options, see the usage page below.
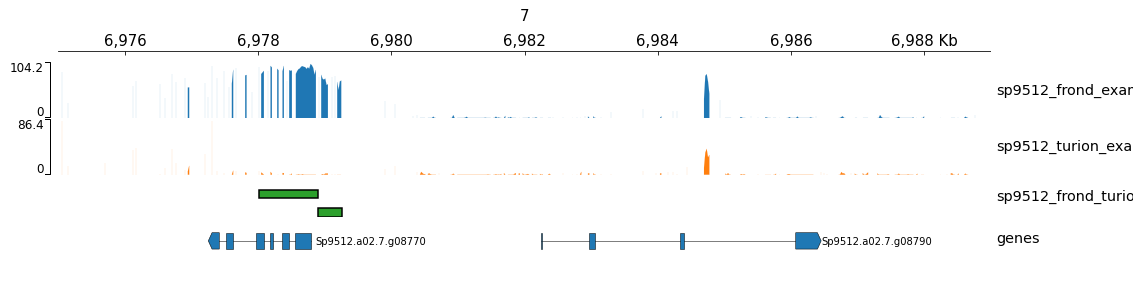
Example with non-human data and BED track
This example command uses data from S. polyrhiza and includes a BED track.
gtracks --genes Sp9512 7:6975000-6989000 sp9512_frond_example.bw sp9512_turion_example.bw sp9512_frond_turion_dmr.bed test-non-human.png
Environment variables
If you want to use your own bigWig files but don't want to write out their
paths every time you run gtracks, you can set your own default tracks using
the environment variable GTRACKS_TRACKS.
export GTRACKS_TRACKS=track1.bw,track2.bw,track3.bw
gtracks output.pdf
You can also change the default gene annotations file and color palette using
environment variables GTRACKS_GENES_PATH and GTRACKS_COLOR_PALETTE.
export GTRACKS_GENES_PATH=path/to/genes.bed.gz
export GTRACKS_COLOR_PALETTE="#color1,#color2,#color3"
gtracks output.pdf
Should your genomic coordinates take a different form from the included default
regex, you may set a different default regex using GTRACKS_COORD_REGEX:
export GTRACKS_COORD_REGEX='[\s\S]+:[0-9]+-[0-9]+$'
Usage
usage: gtracks [-h] [--genes <{path/to/genes.bed.gz,GRCh37,GRCh38,hg19,hg38,Sp9512}>]
[--color-palette <#color> [<#color> ...]] [--max <float>] [--tmp-dir <temp/file/dir>] [--width <int>]
[--genes-height <int>] [--gene-rows <int>] [--x-axis {top,bottom,none}]
[--vlines-bed <path/to/vlines.bed>] [--bed-labels]
<{chr:start-end,GENE}> [<track.{bw,bed}> [<track.{bw,bed}> ...]] <path/to/output.{pdf,png,svg}>
Plot bigWig, bedGraph, and BED signal tracks with gene annotations in a genomic region
positional arguments:
<{chr:start-end,GENE}>
coordinates or gene name to plot
<track.{bw,bdg,bed}> bigWig, bedGraph, or bed files containing tracks
<path/to/output.{pdf,png,svg}>
path to output file
optional arguments:
-h, --help show this help message and exit
--genes <{path/to/genes.bed.gz,GRCh37,GRCh38,hg19,hg38,Sp9512}>
compressed 6-column BED file or 12-column BED12 file containing gene annotations. Alternatively,
providing a genome identifier will use one of the included gene tracks. (default: GRCh37)
--flank <int> add flanks to the plotting region
--color-palette <#color> [<#color> ...]
color pallete for tracks
--max <float> max value of y-axis
--tmp-dir <temp/file/dir>
directory for temporary files
--width <int> width of plot in cm (default: 40)
--genes-height <int> height of genes track (default: 2)
--gene-rows <int> number of gene rows (default: 1)
--x-axis {top,bottom,none}
where to draw the x-axis (default: top)
--vlines-bed <path/to/vlines.bed>
BED file defining vertical lines
--bed-labels include labels on BED tracks
--coord-regex <regex>
regular expression indicating the format for coordinates (default: ([Cc]hr)?[0-9XY]+:[0-9]+-[0-9]+$)
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file gtracks-1.12.6.tar.gz.
File metadata
- Download URL: gtracks-1.12.6.tar.gz
- Upload date:
- Size: 8.4 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.10.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
2adc873c91bdd2933dcd6c528858e0ccc51edf1ca1c952c6a86d131e56adff04
|
|
| MD5 |
c73afd6313019325a3a87147209d865b
|
|
| BLAKE2b-256 |
0917ccb6dcb026cefd8126ed889eb618ec6967e0b60e6b3e1ca7b2d02ea5743c
|
File details
Details for the file gtracks-1.12.6-py3-none-any.whl.
File metadata
- Download URL: gtracks-1.12.6-py3-none-any.whl
- Upload date:
- Size: 8.4 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.10.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
bf590a1eecea539f56a0c7f7f9e8852214d3f19113214f9126b77f08190c376c
|
|
| MD5 |
051537e07e601ef259fba88bab4cfd83
|
|
| BLAKE2b-256 |
b5d861fbfa0a3f24be9d1a3c405d56dec9c77b53ae2804e6efec618ea2df6bf7
|









