Genome-level presence inference from metaproteomic peptide lists.

Project description
MetaUmbra

Genome-level presence inference from metaproteomic peptides
MetaUmbra performs genome-level presence inference from metaproteomic peptide lists. It combines sample-depth-aware genome-unique peptide support with weighted shared peptide evidence to identify statistically supported microbial genomes and generate interpretable presence rankings.
Main features
- Evaluate candidate genome support from metaproteomic peptide tables
- Build genome-specific theoretical peptide references from protein FASTA files
- Support user-defined genome collections, including isolate genomes, strain panels, and MAG catalogs
- Use both unique and shared peptide evidence for genome presence inference
- Calibrate genome-unique peptide counts against sample-specific weak-background genomes by default
- Score multi-sample inputs per sample or user-defined analysis unit without repeated genome digest scans
- Report genome-level p-values, BH-adjusted q-values, and presence scores
- Provide GUI, command-line, and Python workflow support
- Support peptide tables from common metaproteomics workflows such as DIA-NN and MaxQuant
Workflow overview
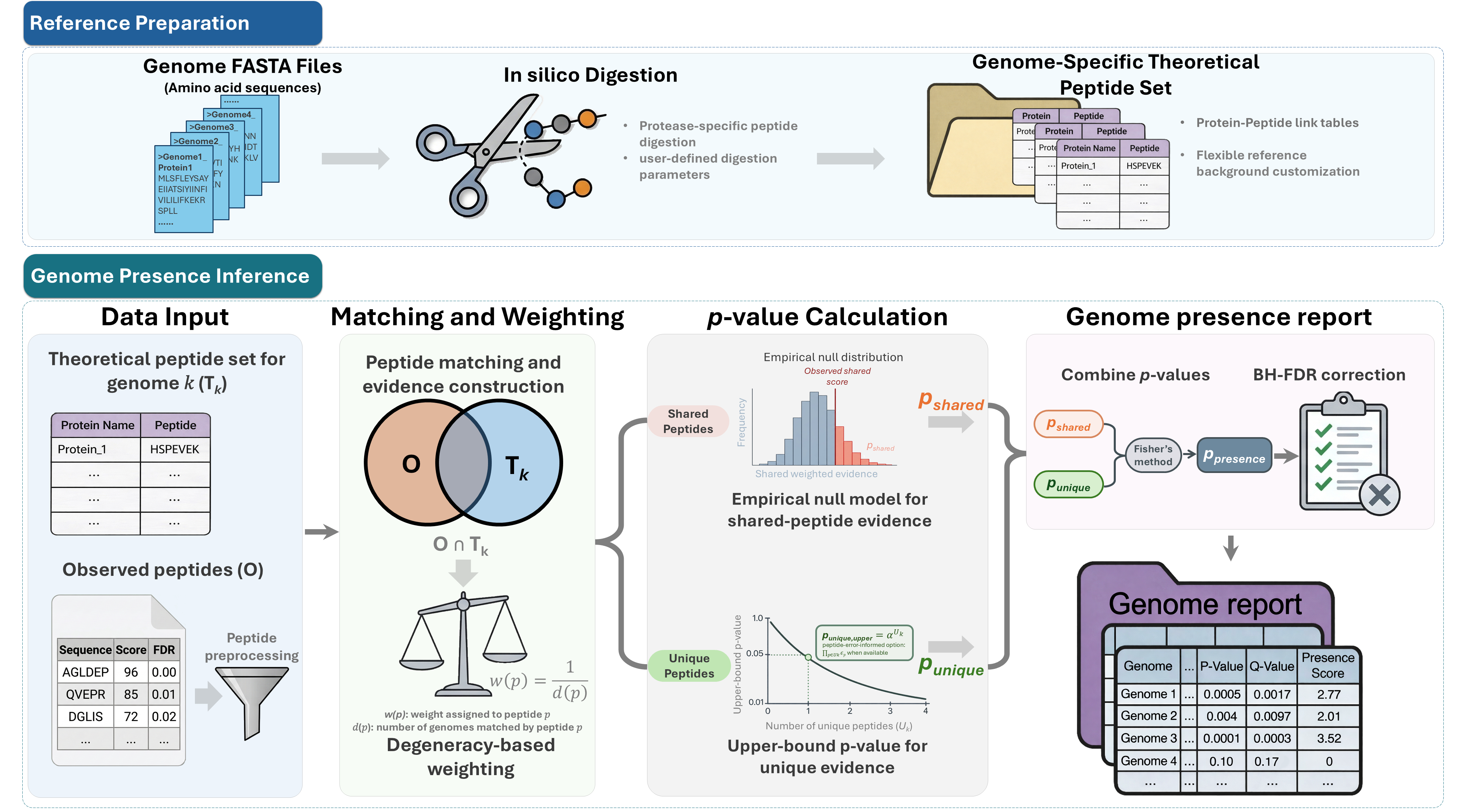
Installation
MetaUmbra requires Python 3.10 or newer. Installation is available via pip from PyPI.
# Install with all features (GUI, parquet support)
pip install metaumbra[all]
The default GUI extra uses PySide6. To run the GUI with PyQt5 instead, install metaumbra[gui-pyqt5].
or
# Install with core features only
pip install metaumbra
Usage
MetaUmbra can be used through either the graphical interface or the command line.
For a detailed walkthrough, including input formats, CLI examples, output interpretation, and troubleshooting, see the MetaUmbra Usage Guide.
Graphical interface
metaumbra-gui
The GUI supports FASTA digestion, peptide table loading, genome presence scoring, and result export.
Command line
MetaUmbra provides separate commands for the main workflow steps:
metaumbra digest --help
metaumbra score --help
metaumbra extract-parquet --help
A typical workflow is:
metaumbra digest ...
metaumbra score ...
Use metaumbra extract-parquet ... to convert DIA-NN parquet reports to peptide TSV files before scoring.
Input
MetaUmbra requires:
- Protein FASTA files, with one FASTA file per genome
- An observed peptide table containing peptide sequences
Optional inputs include peptide scores, peptide-level error values, decoy flags, and genome lineage annotations.
For multi-sample long tables such as DIA-NN reports, metaumbra score --unit-aware can call peptide presence per raw sample using Precursor.Quantity, aggregate samples into analysis_unit_id groups from an optional metadata table, and export clean per-unit, cohort recurrence, sample mapping, unit call-count, and downstream genome-list outputs by default.
Output
The default output is a concise TSV table containing genome-level evidence and significance values. When unit-aware scoring is enabled, the requested output path contains the main unit-level genome presence result and uses a concise downstream-ready schema.
Default unit-aware output files:
<requested output>.tsv: one row peranalysis_unit_idxgenome_id, including unit-specificqvalueandpass_qflags.<stem>_cohort_genome_summary.tsv: one row per genome, summarizing recurrence across units.<stem>_artifacts/unit_aware/<stem>_sample_unit_mapping.tsv: final sample-to-analysis-unit mapping used for the run.<stem>_artifacts/unit_aware/unit_call_counts.tsv: minimal per-unit QC counts.<stem>_artifacts/unit_aware/unit_specific_genome_list_q005.tsv: preferred downstream genome-list interface.<stem>_artifacts/unit_aware/unit_specific_genome_list_q001.tsv: stricter downstream genome-list interface.<stem>_artifacts/unit_aware/unit_aware_manifest.json: compact machine-readable downstream manifest.
The unit-aware TSV files are the canonical tabular outputs. unit_aware_manifest.json is a lightweight downstream integration file for annotation workflows, including MetaX: it maps each analysis_unit_id to sample columns, q<=0.05 genome IDs, and the stricter q<=0.01 subset. It does not duplicate q-values, ranks, lineage, p-values, scores, or peptide counts; those remain in the TSV files. The manifest's default_genome_threshold is q0.05.
Each scoring run creates <stem>_artifacts/ at startup and records run_parameters.json, run.log, run_status.json, and run_summary.json for reproducibility and debugging. The parameter snapshot includes CPU model, logical CPU count, total memory, and platform/architecture metadata. Use --export-diagnostics to write heavier audit and figure-generation outputs such as full_internal_metrics.tsv, knockoff diagnostics, pooled unit-aware results, redundant unit-level subsets, genome unions, and genome-by-unit matrices. Full unit-aware audit columns are written to unit_aware/unit_genome_presence_full.tsv with --export-diagnostics or unit-aware --return-full-table. In the CLI, use --save-cache to write matched_peptides.pkl; in the GUI, the matching "Save matched-peptide cache" option is enabled by default. When cache reuse is requested, the default matched_peptides.pkl cache is preserved during startup cleanup.
In the current implementation, peptide presence within an analysis unit is defined as the union of sample-level peptide presence across samples assigned to that unit. Unit-level p-values combine per-unit shared knockoff evidence (pvalue_shared) with the selected per-unit unique-evidence model (pvalue_unique) using Fisher's method, then apply BH correction separately within each analysis unit.
Key output columns include:
| Column | Description |
|---|---|
genome_id |
Candidate genome identifier |
num_peptides_matched |
Number of observed peptides matched to the genome |
num_peptides_unique |
Number of matched peptides unique to the genome |
theoretical_unique_peptides |
Theoretical peptides unique to this genome among the analyzed genome set, included for hypergeometric-opportunity output |
expected_unique_null |
Expected observed genome-unique peptides under the selected unique-evidence null |
unique_depth_fold |
Observed unique peptides divided by expected unique peptides under the null |
has_unique_evidence |
Whether the genome has at least one observed unique peptide |
pvalue_shared |
Shared-peptide knockoff p-value |
pvalue_unique |
Unique-evidence p-value |
pvalue |
Genome-level p-value |
qvalue |
BH-adjusted genome-level q-value |
presence_score |
Ranking score based on q-value |
Citation
If you use MetaUmbra, please cite:
Wu Q, Ning Z, Zhang A, Cheng K, Figeys D. MetaUmbra: Statistically Controlled Genome-Level Presence Inference from Metaproteomic Peptides.[J]. bioRxiv, 2026.04.29.721689.
A formal citation will be added after publication.
Contact
For questions or issues, please use the GitHub issue tracker or contact the corresponding author listed in the associated manuscript.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file metaumbra-1.3.6.tar.gz.
File metadata
- Download URL: metaumbra-1.3.6.tar.gz
- Upload date:
- Size: 3.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
179505aacf871a64338bf79248aded92eecf06b75ea9aa354e142a2a07844c5b
|
|
| MD5 |
0292653e1fc2ff173b5b3cbfa8ea9c1d
|
|
| BLAKE2b-256 |
ab4815ee028ff2327b395a85579ae4b6c863e5935f0e2ca9279c8567b0484f50
|
File details
Details for the file metaumbra-1.3.6-py3-none-any.whl.
File metadata
- Download URL: metaumbra-1.3.6-py3-none-any.whl
- Upload date:
- Size: 3.3 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a56d69ac58127afc9c623570d1bcfa90d48a06a9f7214f5044cee8a0ec78074b
|
|
| MD5 |
f6a8c7d137b46fc50dbe4928047e756e
|
|
| BLAKE2b-256 |
197f1bf12cec006a7c7c501cfa6e5fd20aece8e8e6bb920ddafb765d8896008a
|







