No project description provided

Project description
MIPMLP
(Microbiome Preprocessing Machine Learning Pipeline)
MIPMLP is a modular pipeline for preprocessing 16S microbiome feature data (ASVs/OTUs) prior to classification tasks using machine learning.
It is based on the paper:
"Microbiome Preprocessing Machine Learning Pipeline", Frontiers in Immunology, 2021 (link)
Background
Raw microbiome data obtained from 16S sequencing (ASVs/OTUs) often requires careful preprocessing before it is suitable for machine learning (ML) classification tasks. MIPMLP (Microbiome Preprocessing Machine Learning Pipeline) was designed to improve ML performance by addressing issues such as sparsity, taxonomic redundancy, and skewed feature distributions.
MIPMLP consists of the following four modular steps:
-
Taxonomy Grouping
Merge features according to a specified taxonomy level: Order, Family, or Genus.
Grouping method options:sum: total abundancemean: average abundancesub-PCA: PCA on each taxonomic group, retaining components explaining ≥50% of the variance
-
Normalization
Normalize feature counts using:log: log10(x + epsilon) — recommendedrelative: divide by total sample counts
-
Standardization (Z-scoring)
Standardize across:- Samples (row-wise)
- Features (column-wise)
- Both
- Or skip standardization altogether
-
Dimensionality Reduction (optional)
Apply PCA or ICA to reduce the number of features.
These steps can be customized via a parameter dictionary as shown below.
How to Use
Installation & Setup
# (optional) create a virtual environment:
python3 -m venv .venv
source .venv/bin/activate # On Windows: .venv\Scripts\activate
# install dependencies:
pip install -r requirements.txt
Run the pipeline
import MIPMLP
# basic usage:
df_processed = MIPMLP.preprocess(df_train)
# full usage:
df_train_processed, df_test_processed = MIPMLP.preprocess(
df_train,
tag=tag_df, # optional
taxonomy_level=7, # default: 7, options: 4-8
taxnomy_group='mean', # default: "mean", options: "sub PCA", "mean", "sum"
epsilon=0.00001, # default: 0.00001, range: 0-1
normalization='log', # default: "log", options: "log", "relative"
z_scoring='No', # default: "No", options: "row", "col", "both", "No"
norm_after_rel='No', # default: "No", options: "No", "z_after_relative" (only used with 'relative')
pca=(0, 'PCA'), # default: (0, "PCA"), use (n, "PCA") for dimensionality reduction, -1 for auto
rare_bacteria_threshold=0.01, # default: 0.01 (1%), removes bacteria that appear in fewer samples
plot=False, # default: False, options: True, False
df_test=df_test_df, # optional: test set to be preprocessed with same parameters
external_sub_pca=sub_pca_model, # optional: use pre-fitted SubPCA model instead of fitting
external_pca=pca_model, # optional: use pre-fitted PCA model instead of fitting
drop_tax_prefix=True # default: True, options: True, False
)
Behavior:
- If
df_testis provided, the pipeline returns both train and test DataFrames . - If not, it returns only the processed train DataFrame.
- You may pass a pre-fitted PCA or SubPCA model; otherwise, the pipeline will fit one for you.
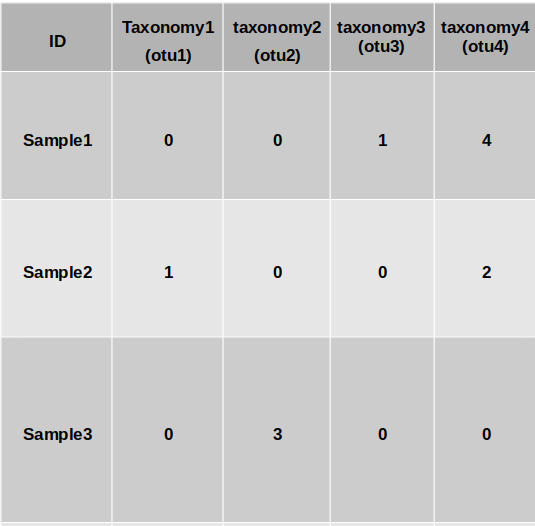
Input Format:
You can provide:
- Option 1: A
.biomfile with raw OTU/ASV counts + a taxonomy.tsvfile - Option 2: A merged
.csvfile that includes both features and taxonomy:- First column:
"ID"(sample IDs) - Rows: individual samples
- Columns: ASVs/features
- Last row: taxonomy info, labeled
"taxonomy"
- First column:
Optional: Tag File
You may also provide a tag file (as a DataFrame) containing class labels for each sample.
This is not required for preprocessing, but if present, MIPMLP will generate additional summary statistics relating features to classes.
Output
The returned value is a preprocessed DataFrame, ready for ML pipelines.
If both train and test are provided, both are returned.
(⚠️ If drop_tax_prefix = True (default), taxonomy prefixes such as k__, p__, g__ will be removed from the feature names. Set this to False if you wish to retain the full taxonomy format in the output table.)
If plot = True , a histogram showing the percentage of samples in which each bacterium appears.
(⚠️ If pca is enabled, plot=True is not recommended. The visualization will not reflect the original features post-dimensionality reduction.)
Example histogram visualization:

iMic
iMic is a method to combine information from different taxa and improves data representation for machine learning using microbial taxonomy. iMic translates the microbiome to images, and convolutional neural networks are then applied to the image.
micro2matrix
Translates the microbiome values and the cladogram into an image. micro2matrix also saves the images that were created in a given folder.
Input
-df A pandas dataframe which is similar to the MIPMLP preprocessing's input (above). -folder A folder to save the new images at.
Parameters
You can determine all the MIPMLP preprocessing parameters too, otherwise it will run with its deafulting parameters (as explained above).
How to use
import pandas as pd
df = pd.read_csv("address/ASVS_file.csv")
folder = "save_img_folder"
MIPMLP.micro2matrix(df, folder)
CNN2 class - optional
A model of 2 convolutional layer followed by 2 fully connected layers.
####CNN model parameters -l1 loss = the coefficient of the L1 loss -weight decay = L2 regularization -lr = learning rate -batch size = as it sounds -activation = activation function one of: "elu", | "relu" | "tanh" -dropout = as it sounds (is common to all the layers) -kernel_size_a = the size of the kernel of the first CNN layer (rows) -kernel_size_b = the size of the kernel of the first CNN layer (columns) -stride = the stride's size of the first CNN -padding = the padding size of the first CNN layer -padding_2 = the padding size of the second CNN layer -kernel_size_a_2 = the size of the kernel of the second CNN layer (rows) -kernel_size_b_2 = the size of the kernel of the second CNN layer (columns) -stride_2 = the stride size of the second CNN -channels = number of channels of the first CNN layer -channels_2 = number of channels of the second CNN layer -linear_dim_divider_1 = the number to divide the original input size to get the number of neurons in the first FCN layer -linear_dim_divider_2 = the number to divide the original input size to get the number of neurons in the second FCN layer -input dim = the dimention of the input image (rows, columns)
How to use
params = {
"l1_loss": 0.1,
"weight_decay": 0.01,
"lr": 0.001,
"batch_size": 128,
"activation": "elu",
"dropout": 0.1,
"kernel_size_a": 4,
"kernel_size_b": 4,
"stride": 2,
"padding": 3,
"padding_2": 0,
"kernel_size_a_2": 2,
"kernel_size_b_2": 7,
"stride_2": 3,
"channels": 3,
"channels_2": 14,
"linear_dim_divider_1": 10,
"linear_dim_divider_2": 6,
"input_dim": (8,100)
}
model = MIPMLP.CNN(params)
A trainer on the model should be applied by the user after choosing the best hyperparameters by an NNI platform.
apply_iMic (a basic example run of iMic function)
A basic running iMic option of uploading the images dividing them to a training set and test set and returns the real labels (train and test) and the predicted labels (train and test)
Input
-tag A tag pandas dataframe with similar samples to the raw ASVs file. -folder A folder of the saved images from the micro2matrix step. -test_size Fraction of the test set from the whole cohort (default is 0.2). -params iMic model's hyperparameters. Should be selected for each dataset separately by grid-search or NNI on appropriate validation set. The default params are { "l1_loss": 0.1, "weight_decay": 0.01, "lr": 0.001, "batch_size": 128, "activation": "elu", "dropout": 0.1, "kernel_size_a": 4, "kernel_size_b": 4, "stride": 2, "padding": 3, "padding_2": 0, "kernel_size_a_2": 2, "kernel_size_b_2": 7, "stride_2": 3, "channels": 3, "channels_2": 14, "linear_dim_divider_1": 10, "linear_dim_divider_2": 6, "input_dim": (8, 235) })
Note that the input_dim is also updated automatically during the run.
Output
A dictionary of {"pred_train": pred_train,"pred_test": pred_test,"y_train": y_train,"y_test": y_test}
How to use
# Load tag
tag = pd.read_csv("data/ibd_tag.csv", index_col=0)
# Prepare iMic images
otu = pd.read_csv("data/ibd_for_process.csv")
MIPMLP.micro2matrix(otu, folder="data/2D_images")
# Run a toy iMic model. One should optimize hyperparameters before
dct = apply_iMic(tag, folder="data/2D_images")
Citation
-
Shtossel, Oshrit, et al. "Ordering taxa in image convolution networks improves microbiome-based machine learning accuracy." Gut Microbes 15.1 (2023): 2224474.
-
Jasner, Yoel, et al. "Microbiome preprocessing machine learning pipeline." Frontiers in Immunology 12 (2021): 677870.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file mipmlp-1.2.19.tar.gz.
File metadata
- Download URL: mipmlp-1.2.19.tar.gz
- Upload date:
- Size: 24.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ef7a71994375d1f624e9371f6de9a9c67b25417e39356769a17b91a085a79b3a
|
|
| MD5 |
f3195ddb8b06ff509d46f559cdf5842f
|
|
| BLAKE2b-256 |
46d65c18400fb7616aae89209c4ad3b4f8610544165e133ce594175664ef1554
|
File details
Details for the file mipmlp-1.2.19-py3-none-any.whl.
File metadata
- Download URL: mipmlp-1.2.19-py3-none-any.whl
- Upload date:
- Size: 29.3 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5dadbd9b2c33b505b4be44c58671682f58df6f26097c1f01b02957ea0638703a
|
|
| MD5 |
6ad90c9043b828e37d8fc962ea603130
|
|
| BLAKE2b-256 |
a24701baa6c0377ec5ee09aa33c9792fb64d26edbe3604a574d89c49b0f2234a
|







