mlmm - ML/MM toolkit for enzyme reaction analysis
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
ML/MM toolkit — Towards Accelerated Mechanistic Investigation of Enzymatic Reactions
Overview
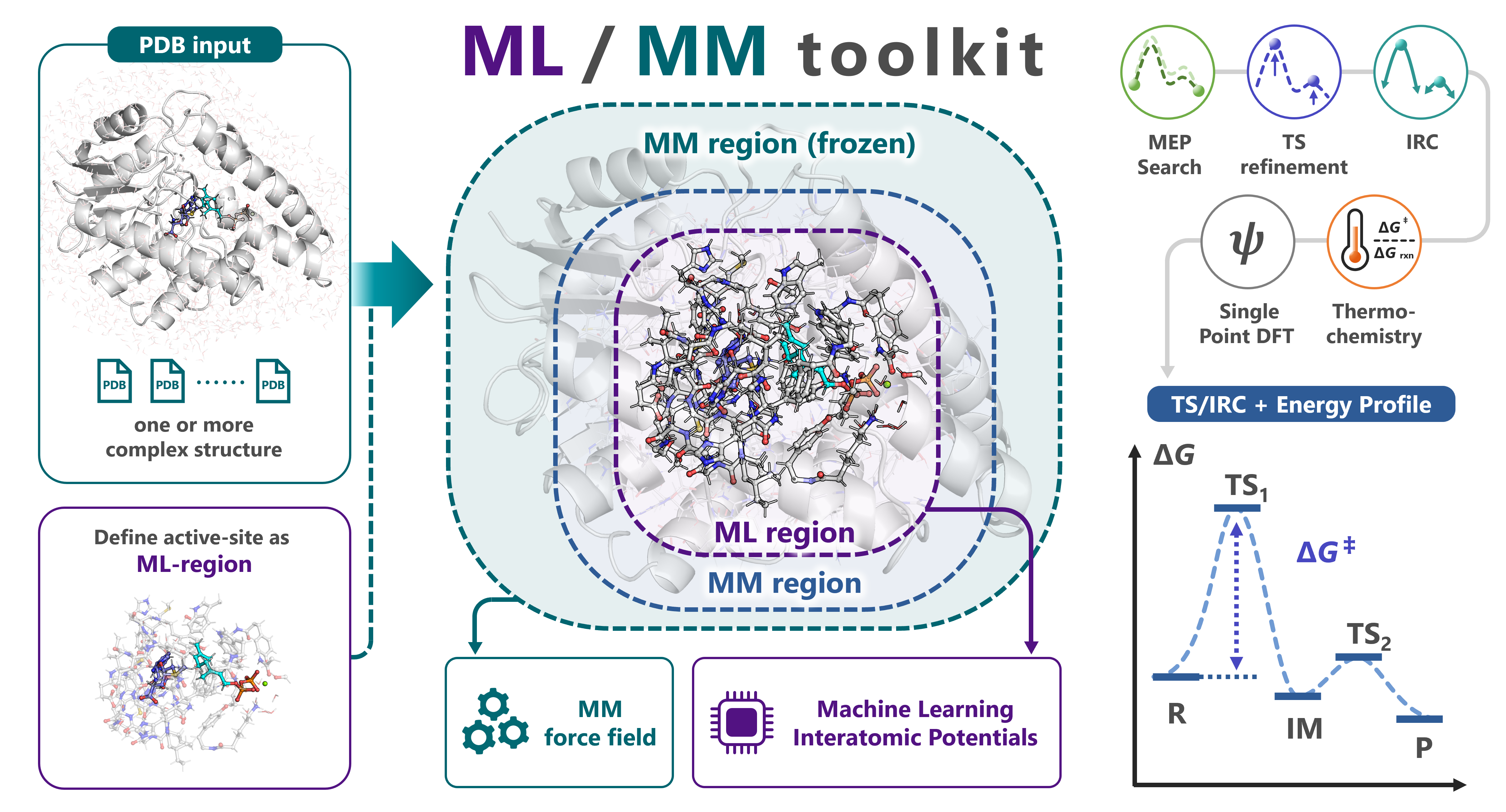
mlmm-toolkit is an open-source CLI for ML/MM ONIOM analyses of enzymatic reactions. It replaces the QM region of conventional QM/MM with a machine-learning interatomic potential (MLIP, default: UMA) while keeping the surrounding protein under an analytical Amber force field (hessian_ff), and chains MM parametrization → ML-region selection → MEP search → TS optimization → IRC → thermochemical correction → DFT single-point in one command. A link-atom boundary handles amino-acid residues straddling the ML/MM cut, and a microiteration scheme makes TS optimization and Hessian-based methods tractable on ~10 000-atom systems.
Test a reaction mechanism in a single command:
# Multi-structure MEP (R + P endpoints → MEP, with TS optimization + thermo)
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermo
For scan-mode on a single structure and the bundled methyltransferase walk-through, see examples/. Each stage is also exposed as an individual subcommand.
Prerequisites: input PDBs must already contain hydrogens; multiple PDBs must share the same atoms in the same order (only coordinates differ).
mlmm allrunsmm-parmautomatically. Match-l RES:CHARGEto the H count actually present (e.g. SAM with 23 H =SAM:1cation, 22 H =SAM:0neutral) — full input-prep checklist in docs/getting-started.md.
Related tools
| Tool | Use case |
|---|---|
| pdb2reaction | Pure-MLIP reaction paths for cluster models and small molecules from PDB / XYZ / GJF. |
| uma_pysis | Lightweight YAML-driven UMA–pysisyphus interface for quick/exploratory reaction-mechanism studies (GS / TS / IRC / ΔG). |
mlmm-toolkitbundles a GPU-optimized pysisyphus fork that is not compatible with upstream pysisyphus — do not install it into an environment that already has upstream pysisyphus.
Documentation
- Getting Started · Concepts · Installation · Troubleshooting
- Python API · CLI Conventions · YAML Reference · JSON Output Schema
- Full command index: docs/index.md
System requirements
| Component | Requirement |
|---|---|
| OS / Python | Linux recommended; native Windows unsupported (AmberTools/tleap unavailable). Python >= 3.11. |
| GPU / CUDA / VRAM | NVIDIA GPU, CUDA >= 12.6 (12.8+ recommended; required for RTX 50-series). 8 GB+ VRAM recommended. |
| RAM / Disk | 32 GB+ RAM recommended; 20 GB free disk for the conda env, AmberTools, UMA cache, and artifacts. |
Required external tools: AmberTools (tleap) and pdbfixer — conda install -c conda-forge ambertools pdbfixer -y. CPU-only execution works for setup commands but is 10–100× slower for any ML/MM dynamics or Hessian step. Full requirement and tuning details: docs/getting-started.md#installation.
Installation
# 1. New env + AmberTools + CUDA-enabled PyTorch
conda create -n mlmm-toolkit python=3.11 -y && conda activate mlmm-toolkit
conda install -c conda-forge ambertools pdbfixer -y
pip install torch --index-url https://download.pytorch.org/whl/cu129
# 2. mlmm-toolkit (editable from a local clone, or `pip install mlmm-toolkit`)
pip install -e .
# 3. Authenticate Hugging Face once (only required for the default UMA backend)
# Accept the FAIR Chemistry License v1 at https://huggingface.co/facebook/UMA, then:
hf auth login # interactive
# OR: export HF_TOKEN=hf_xxx && hf auth login --token "$HF_TOKEN" # CI / HPC
Avoid AmberTools conflicts: on clusters with a system AmberTools module loaded, run
module unload amberbefore installing to prevent a ParmEd conflict with the conda-installed AmberTools.
Optional extras (install only what you need):
| Extra | Adds |
|---|---|
[orb] / [aimnet] |
Orb / AIMNet2 MLIP backend — not HF-gated |
[dft] |
PySCF + GPU4PySCF single-point DFT (--dft / mlmm dft) — practical to ~500 ML-region atoms |
[mcp] |
Model Context Protocol server (mlmm-mcp) for agent clients |
[pdbfixer] |
PDBFixer extra (alternative to the conda install above) |
The MACE backend (-b mace) is not a pip extra: mace-torch pins e3nn==0.4.4, which conflicts with fairchem-core's e3nn>=0.5 (UMA), so it needs a dedicated environment — pip uninstall -y fairchem-core && pip install mace-torch (see docs/getting-started.md#installation).
CUDA module-load recipes, alternative-backend installs, DMF / cyipopt, Plotly Chromium, and HPC job-script templates: docs/getting-started.md and docs/device-hpc.md.
Preparing an Enzyme-Substrate System
For most systems the only hard requirement is a PDB with explicit hydrogens (at the intended protonation state). mlmm all then builds the MM topology, selects the ML region, and runs the whole pipeline in one command — see Quick Examples for the three input modes (multi-structure R → P, single-structure scan, TS-only). The preparation steps below are optional.
-
Build a structural model of the complex. Download coordinates from the Protein Data Bank. If an experimental structure is not available, use structure-prediction programs such as AlphaFold3, Boltz2, or Chai; docking programs; or GUI software such as PyMOL. Add hydrogens at the intended protonation state (or let
mm-parm --add-h --ph 7add them). For multi-structure (R → P) runs, every PDB must share the same atoms in the same order. -
(Optional) Build the MM topology yourself — it is automatic by default.
mlmm all(viamlmm mm-parm) generates the Amber.parm7/.rst7from the PDB automatically; unknown residues (ligands, cofactors) are parameterized with GAFF2 / AM1-BCC — pass formal charges with-l 'RES:CHARGE'. Build the topology by hand when it helps — a custom force field, special solvation, or a system the automatic route cannot handle — then pass it with--parm. To mimic aqueous conditions, solvate the complex and remove water molecules beyond ~6 Å (see the OpenMM cookbook / tleap).Note: elemental information (columns 77–78) is omitted in PDB files generated by tleap. Use
mlmm add-elem-infoto fix this. -
(Optional) Define the ML region yourself.
mlmm allextracts the ML region from-c/--centerand-r/--radiusautomatically. To define it yourself instead, build an ML-region PDB — withmlmm extractor any molecular viewer — and feed it tomlmm all(or the per-stage subcommands) with--model-pdb; this skips the automatic extraction:mlmm extract -i complex.pdb -c 'SAM,GPP' -r 6.0 -l 'SAM:1,GPP:-3' -o ml_region.pdb
Important: atom order, residue names, and residue numbers must match between the full PDB and the ML-region PDB. (In PyMOL, tick "Original atom order" when exporting.)
Quick Examples
# Multi-structure MEP (R + P → MEP, with TS + thermo + DFT)
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermo --dft
# Scan mode (single structure → staged bond scans → MEP)
mlmm all -i R.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' \
--scan-lists "[('SAM 359 CS1','GPP 360 C8',1.3)]"
# TS-only validation (existing TS candidate)
mlmm tsopt -i TS_candidate_layered.pdb --parm complex.parm7 -q 1 --opt-mode grad
For Gaussian-ONIOM / ORCA-QM/MM round-trips use oniom-export / oniom-import. Per-stage walkthrough (mm-parm → extract → define-layer → opt → path-search → tsopt → freq → irc → dft): docs/getting-started.md and docs/quickstart-all.md. Working scripts (methyltransferase + toy_system): examples/.
Output
A run writes its deliverables to --out-dir (default ./result_all/):
segments/seg_NN/{reactant,ts,product}.pdb— the canonical R / TS / P structures to citemep.pdb/mep_trj.xyz— the merged reaction path;energy_diagram_MEP.png— barrier diagramsummary.log(human-readable) /summary.json(machine-readable)- Reusable inputs for follow-up runs:
ml_region.pdb(--model-pdb),mm_parm/*.parm7(--parm),layered/(B-factor-annotated full-system PDBs)
Pipeline scratch lives under _work/ (safe to delete). Full layout and filename conventions: docs/output-layout.md.
CLI Subcommands
| Subcommand | Role | Doc |
|---|---|---|
all (default) |
End-to-end: mm-parm → extract → MEP → TS → IRC → freq → DFT |
all |
mm-parm |
Generate parm7/rst7 via AmberTools | mm-parm |
extract |
Extract active-site pocket | extract |
define-layer |
Assign 3-layer ML/MM B-factor encoding | define-layer |
add-elem-info / fix-altloc |
Repair PDB element columns / resolve altlocs | add-elem-info · fix-altloc |
opt / tsopt |
Geometry / TS optimization | opt · tsopt |
path-opt / path-search |
MEP via GSM/DMF; recursive refinement | path-opt · path-search |
scan / scan2d / scan3d |
1D / 2D / 3D bond-distance scans | scan · scan2d · scan3d |
freq / irc |
Vibrational analysis + thermo / IRC (EulerPC) | freq · irc |
dft / sp |
Single-point DFT / single-point ML/MM ONIOM | dft · sp |
bond-summary |
Compare structures, report bond changes | bond-summary |
trj2fig / energy-diagram |
Energy plot / R→TS→P diagram | trj2fig · energy-diagram |
oniom-export / oniom-import |
Gaussian ONIOM / ORCA QM/MM round-trip | oniom-export · oniom-import |
3-layer system (ML / Movable-MM / Frozen-MM, B-factor encoded), link-atom treatment, units (eV·Å in core / Ha·Bohr in pysisyphus CLI): docs/concepts.md. Python API (MLMMCore, MLMMASECalculator, pysisyphus mlmm calculator): docs/python-api.md.
Getting Help
mlmm --help # top-level
mlmm <subcmd> --help # core options
mlmm <subcmd> --help-advanced # full option set
Issues: https://github.com/t-0hmura/mlmm_toolkit/issues.
Citation
@article{ohmura2025mlmm,
author = {Ohmura, Takuto and Inoue, Sei and Terada, Tohru},
title = {ML/MM Toolkit -- Towards Accelerated Mechanistic Investigation of Enzymatic Reactions},
year = {2025}, journal = {ChemRxiv}, doi = {10.26434/chemrxiv-2025-jft1k}
}
Agent Skills
Agent Skills for Claude Code / Codex / Cursor etc. in skills/ — copy into your project's skill location (e.g. .claude/skills/) to let an agent drive mlmm-toolkit workflows and subcommands.
Known limitations
- MACE + UMA cannot coexist (
e3nnversion conflict). Use separate conda envs. - DFT single-point is practical to ~500 ML-region atoms; larger regions incur high computational cost.
- ORB backend sometimes converges TS with extra soft imaginary modes — prefer UMA / MACE for clean single-saddle spectra.
- CPU-only execution is 10–100× slower than GPU; AmberTools (
tleap) is required formm-parm.
Contributing
Issues and pull requests are welcome — see CONTRIBUTING.md.
License
GNU General Public License v3 (GPL-3.0).
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file mlmm_toolkit-0.3.0.tar.gz.
File metadata
- Download URL: mlmm_toolkit-0.3.0.tar.gz
- Upload date:
- Size: 5.8 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ea60d87b6e0561063b8cbb40e2e3645a5420966ce075b4e35e6b50148dd52f4d
|
|
| MD5 |
bd456a56a0d30a9ab749b34075c6cfae
|
|
| BLAKE2b-256 |
d4b924bf9555bc2ec75c2096e901f25ed1d945952bc1f01aa73b0860137c3b9c
|
Provenance
The following attestation bundles were made for mlmm_toolkit-0.3.0.tar.gz:
Publisher:
release.yml on t-0hmura/mlmm_toolkit
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
mlmm_toolkit-0.3.0.tar.gz -
Subject digest:
ea60d87b6e0561063b8cbb40e2e3645a5420966ce075b4e35e6b50148dd52f4d - Sigstore transparency entry: 2017548900
- Sigstore integration time:
-
Permalink:
t-0hmura/mlmm_toolkit@90e303ca4c3563e8f70b3edf4c86fd327339e612 -
Branch / Tag:
refs/tags/v0.3.0 - Owner: https://github.com/t-0hmura
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@90e303ca4c3563e8f70b3edf4c86fd327339e612 -
Trigger Event:
release
-
Statement type:
File details
Details for the file mlmm_toolkit-0.3.0-py3-none-any.whl.
File metadata
- Download URL: mlmm_toolkit-0.3.0-py3-none-any.whl
- Upload date:
- Size: 1.0 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e383cfee5d6aa305ad60beb2c2aaf123b4e63f8aad651f721f0424fd77197cea
|
|
| MD5 |
3dcfcf7dc2d2edb662ecc6bf4156b067
|
|
| BLAKE2b-256 |
b0fd0317edfa83e525f281fdfb36a82e16212e4d06df60bad89a45da830d8956
|
Provenance
The following attestation bundles were made for mlmm_toolkit-0.3.0-py3-none-any.whl:
Publisher:
release.yml on t-0hmura/mlmm_toolkit
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
mlmm_toolkit-0.3.0-py3-none-any.whl -
Subject digest:
e383cfee5d6aa305ad60beb2c2aaf123b4e63f8aad651f721f0424fd77197cea - Sigstore transparency entry: 2017549012
- Sigstore integration time:
-
Permalink:
t-0hmura/mlmm_toolkit@90e303ca4c3563e8f70b3edf4c86fd327339e612 -
Branch / Tag:
refs/tags/v0.3.0 - Owner: https://github.com/t-0hmura
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@90e303ca4c3563e8f70b3edf4c86fd327339e612 -
Trigger Event:
release
-
Statement type:







