A lightweight and easy-to-use Python bioinformatics toolkit.

Project description
omiBio
- A Lightweight Bioinformatics Toolkit





Introduction
omiBio is a lightweight, user-friendly Python toolkit for bioinformatics — ideal for education, research, and rapid prototyping.
Key features:
- Robust data structures:
Sequence,Polypeptide, etc., with optional validation. - Simple I/O: Read/write bioinformatics files (e.g., FASTA) with one-liners.
- Analysis tools: GC content, ORF detection, consensus sequences, sliding windows, and more.
- CLI included: Run common tasks from the terminal .
- Basic visualization: Built-in plotting (via matplotlib & seaborn) for quick insights.
- Functional & OOP APIs: Use classes or convenient wrapper functions.
Modules Overview
The omiBio toolkit is organized into the following modules:
| Module | Purpose | Key Classes / Functions |
|---|---|---|
omibio.sequence |
Sequence-type data structures | Sequence, Polypeptide |
omibio.bio |
Biological objects and data containers | SeqInterval, AnalysisResult |
omibio.io |
File I/O for common bioinformatics formats | read_fasta(), read_fastq() |
omibio.analysis |
Sequence analysis functions | gc(), sliding_gc(), find_orfs() |
omibio.utils |
General-purpose utility functions | truncate_repr() |
omibio.viz |
Simple and easy-to-use data visualization | plot_orf(), plot_sliding_gc() |
omibio.cli |
Command-line interfaces for common workflows | omibio random-fasta, omibio clean |
Release Notes - omiBio [v0.1.4] 12/14/25
Performance & Core I/O
-
Optimized FASTA parsing
Introduced the generator-basedread_fasta_iter()to improve performance, refine error handling, and add a configurable warning system.
The existingread_fasta()API remains unchanged for external use and continues to returnSeqCollections, allowing users to choose between eager and lazy parsing.
Bothread_fasta()andread_fasta_iter()now acceptTextIOandPathLikeobjects as data sources. -
FASTQ support
Addedread_fastq()andwrite_fastq()with the same design philosophy as the FASTA APIs.
A generator interface,read_fastq_iter(), is also provided.
All FASTQ I/O functions supportTextIOandPathLikeinputs. -
Flexible file writing
All sequence writing functions can now return a list of formatted strings when no output file is specified.
CLI Improvements
- Refactored and streamlined the CLI structure.
- Improved existing commands and added new ones, including:
omibio fasta viewomibio fastq to-fastaomibio kmer count
- All CLI commands support
stdin/stdoutand can be composed in Unix-style pipelines.
API & Data Model Changes
- Removed the
GeneandGenomeclasses, which overlapped in functionality withSeqEntryandSeqCollections. - Made the
SequenceandPolypeptideclasses immutable. - Added the
at_content()method to theSequenceclass. - Applied
__slots__toSeqIntervalandSeqEntryto reduce memory overhead.
Analysis & Visualization
- Enhanced
plot_kmer()to support k-mer heatmaps across multiple sequences. - Refactored
AnalysisResultinto an abstract base class. - Added concrete result types:
IntervalResultKmerResult
- Results returned by analysis functions (e.g.
kmer()) can now be visualized directly via a unified.plot()interface.
Quality Assurance
- Numerous minor fixes and internal refinements.
- Comprehensive test coverage (≥ 95%).
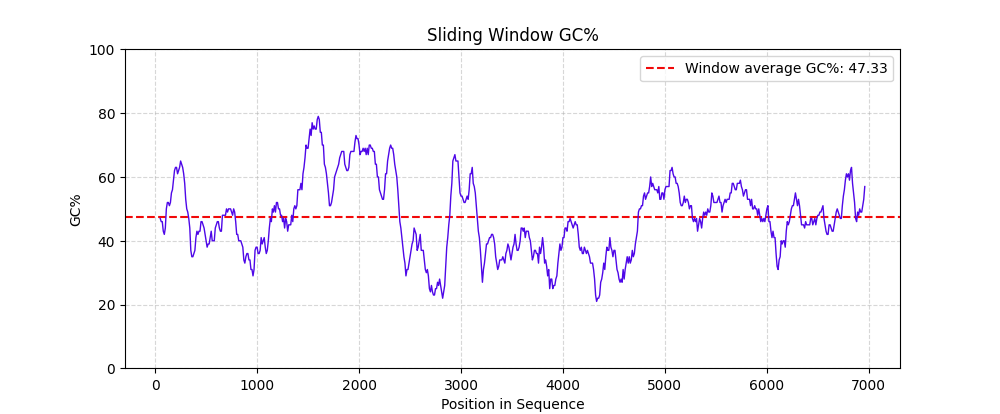
Usage example
Creating a sliding window GC chart using omiBio:
# Load sequences from FASTA (returns dict[str, Sequence])
seqs: SeqCollections[SeqEntry] = read_fasta("examples/example.fasta")
dna: Sequence = seqs["example"]
# Compute GC content in sliding windows (window=200 bp, step=20 bp)
result: IntervalResult[SeqInterval] = sliding_gc(dna, window=200, step=20)
# Visualize easily
result.plot(show=True) # or: plot_sliding_gc(result, show=True)
Or even a one-liner:
sliding_gc(read_fasta("examples/example.fasta")["example"]).plot(show=True)
The above code will produce results like this:
Using omiBio's Command-line interfaces:
$ omibio orf find example.fasta --min-length 100
The above CLI will produce results like this:
seq_id start end strand frame length
example_2 70 289 - -2 219
example_16 53 257 + +3 204
example_13 118 301 + +2 183
example_4 92 272 - -1 180
example_2 157 322 + +2 165
example_5 17 173 - -1 156
example_16 176 332 - -1 156
...
Installation / 安装
From PyPI:
$ pip install omibio
Requirements
- Python: >= 3.12
- Core dependencies:
click(for CLI)numpy&pandas→ analysis/plotting dependenciesmatplotlib&seaborn→ enables visualization
For complete project build and dependency configuration, please refer to pyproject.toml
Code Style
omiBio follows PEP 8 conventions for Python code.
All code is automatically formatted and checked using flake8.
License
This project is licensed under the MIT License.
Things to note
- Most of the code in this project uses 0-based indexes, half-open interval, rather than the 1-based indexes commonly used in biology.
- All code type hints in this project use PEP 585 generic syntax in Python 3.9+.
- This project is still under development and not yet ready for production. Please use it with caution. If you have any suggestions, please contact us:
- gmail: linkaiwen048@gmail.com
- qq: 2658592119
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file omibio-0.1.4.post2.tar.gz.
File metadata
- Download URL: omibio-0.1.4.post2.tar.gz
- Upload date:
- Size: 42.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ea4b4aba7cd0282251d8b0fcd65353b70b40ebda6166d516ddc269574d4f0acc
|
|
| MD5 |
2b41c7e17198634fac303e6645257125
|
|
| BLAKE2b-256 |
a9459d473b5c7d09b24f98b232e62ddeab24b102fe1ea42357a2f1a8e040ce82
|
Provenance
The following attestation bundles were made for omibio-0.1.4.post2.tar.gz:
Publisher:
python-publish.yml on LK923/omiBioKit
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
omibio-0.1.4.post2.tar.gz -
Subject digest:
ea4b4aba7cd0282251d8b0fcd65353b70b40ebda6166d516ddc269574d4f0acc - Sigstore transparency entry: 763985257
- Sigstore integration time:
-
Permalink:
LK923/omiBioKit@734e501b0da97d88efa63444d308620696516623 -
Branch / Tag:
refs/tags/v0.1.4.post2 - Owner: https://github.com/LK923
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
python-publish.yml@734e501b0da97d88efa63444d308620696516623 -
Trigger Event:
release
-
Statement type:
File details
Details for the file omibio-0.1.4.post2-py3-none-any.whl.
File metadata
- Download URL: omibio-0.1.4.post2-py3-none-any.whl
- Upload date:
- Size: 65.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
542f4434b8db87fd8ab6bf22df1175b29e6d48aad69d496ec727b9f2b5e5746c
|
|
| MD5 |
35d47bd498e20b505bfcde0ca7ffafd6
|
|
| BLAKE2b-256 |
c875889ec5f37dab827a161645ba1a4770df2ae6453add990d43caaef5c4b3c9
|
Provenance
The following attestation bundles were made for omibio-0.1.4.post2-py3-none-any.whl:
Publisher:
python-publish.yml on LK923/omiBioKit
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
omibio-0.1.4.post2-py3-none-any.whl -
Subject digest:
542f4434b8db87fd8ab6bf22df1175b29e6d48aad69d496ec727b9f2b5e5746c - Sigstore transparency entry: 763985258
- Sigstore integration time:
-
Permalink:
LK923/omiBioKit@734e501b0da97d88efa63444d308620696516623 -
Branch / Tag:
refs/tags/v0.1.4.post2 - Owner: https://github.com/LK923
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
python-publish.yml@734e501b0da97d88efa63444d308620696516623 -
Trigger Event:
release
-
Statement type:







