Unofficial fork of ProteinMPNN (Dauparas et al., Science 2022) with Apple Silicon (MPS) support
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
ProteinMPNN (MPS Fork)
Unofficial fork of dauparas/ProteinMPNN with Apple Silicon (MPS) support, packaged for
pip install. Original work © Justas Dauparas et al., Baker Lab. Released under MIT.
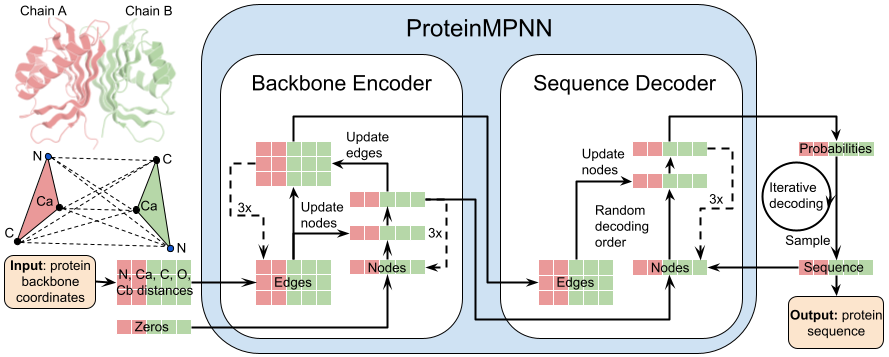
Install
# From PyPI
pip install proteinmpnn-mps
# Latest dev (from this fork)
pip install git+https://github.com/tmsincomb/ProteinMPNN.git
# With helper-script extras (Biopython, for cif_to_pdb.py)
pip install "proteinmpnn-mps[helpers]"
# With training extras (pandas, psutil)
pip install "proteinmpnn-mps[training]"
After install, the runner is available as a console command:
proteinmpnn --pdb_path input.pdb --out_folder ./out --num_seq_per_target 2
It also still works as a script: python protein_mpnn_run.py ....
Apple Silicon (MPS)
This fork auto-selects the MPS backend on Apple Silicon Macs (CUDA > MPS > CPU). Requires PyTorch ≥ 1.12 and macOS ≥ 12.3. See docs/mps/ProteinMPNN_MPS_Notes.md for performance notes, environment variables, and known gotchas.
Releasing a new version (maintainer)
Releases are published to PyPI via GitHub Actions (.github/workflows/publish.yml) using PyPI Trusted Publishing (OIDC) — no API tokens stored in the repo.
One-time setup:
- Register
proteinmpnn-mpson PyPI (upload first build manually, or pre-register). - On PyPI → project → Settings → Publishing → Add a new pending publisher:
- Owner:
tmsincomb - Repository:
ProteinMPNN - Workflow:
publish.yml - Environment:
pypi
- Owner:
- In GitHub repo Settings → Environments → New environment named
pypi.
Per-release:
# bump version in pyproject.toml, commit, then:
git tag v1.0.1.mps1
git push origin v1.0.1.mps1
Tag push triggers the workflow → builds sdist+wheel → publishes to PyPI.
Manual conda install (alternative)
To run ProteinMPNN clone this github repo and install Python>=3.0, PyTorch, Numpy.
Full protein backbone models: vanilla_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt, v_48_030.pt, soluble_model_weights/v_48_010.pt, v_48_020.pt.
CA only models: ca_model_weights/v_48_002.pt, v_48_010.pt, v_48_020.pt. Enable flag --ca_only to use these models.
Helper scripts: helper_scripts - helper functions to parse PDBs, assign which chains to design, which residues to fix, adding AA bias, tying residues etc.
Code organization:
protein_mpnn_run.py- the main script to initialialize and run the model.protein_mpnn_utils.py- utility functions for the main script.examples/- simple code examples.inputs/- input PDB files for examplesoutputs/- outputs from examplescolab_notebooks/- Google Colab examplestraining/- code and data to retrain the model
Input flags for protein_mpnn_run.py:
argparser.add_argument("--suppress_print", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--ca_only", action="store_true", default=False, help="Parse CA-only structures and use CA-only models (default: false)")
argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;")
argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise")
argparser.add_argument("--use_soluble_model", action="store_true", default=False, help="Flag to load ProteinMPNN weights trained on soluble proteins only.")
argparser.add_argument("--seed", type=int, default=0, help="If set to 0 then a random seed will be picked;")
argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-log_prob to npy files")
argparser.add_argument("--path_to_fasta", type=str, default="", help="score provided input sequence in a fasta format; e.g. GGGGGG/PPPPS/WWW for chains A, B, C sorted alphabetically and separated by /")
argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position")
argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs")
argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)")
argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)")
argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output unconditional probabilities p(s_i given backbone) in one forward pass")
argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms")
argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target")
argparser.add_argument("--batch_size", type=int, default=1, help="Batch size; can set higher for titan, quadro GPUs, reduce this if running out of GPU memory")
argparser.add_argument("--max_length", type=int, default=200000, help="Max sequence length")
argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.2 0.25 0.5. Sampling temperature for amino acids. Suggested values 0.1, 0.15, 0.2, 0.25, 0.3. Higher values will lead to more diversity.")
argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/")
argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed")
argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB ")
argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed pdb into jsonl")
argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.")
argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions")
argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.")
argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias if neededi, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.")
argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.")
argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices")
argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm")
argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions")
argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs")
argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions")
For example to make a conda environment to run ProteinMPNN:
conda create --name mlfold- this creates conda environment calledmlfoldsource activate mlfold- this activate environmentconda install pytorch torchvision torchaudio cudatoolkit=11.3 -c pytorch- install pytorch following steps from https://pytorch.org/
These are provided examples/:
submit_example_1.sh- simple monomer examplesubmit_example_2.sh- simple multi-chain examplesubmit_example_3.sh- directly from the .pdb pathsubmit_example_3_score_only.sh- return score only (model's uncertainty)submit_example_3_score_only_from_fasta.sh- return score only (model's uncertainty) loading sequence from fasta filessubmit_example_4.sh- fix some residue positionssubmit_example_4_non_fixed.sh- specify which positions to designsubmit_example_5.sh- tie some positions together (symmetry)submit_example_6.sh- homooligomer examplesubmit_example_7.sh- return sequence unconditional probabilities (PSSM like)submit_example_8.sh- add amino acid biassubmit_example_pssm.sh- use PSSM bias when designing sequences
Output example:
>3HTN, score=1.1705, global_score=1.2045, fixed_chains=['B'], designed_chains=['A', 'C'], model_name=v_48_020, git_hash=015ff820b9b5741ead6ba6795258f35a9c15e94b, seed=37
NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER/NMYSYKKIGNKYIVSINNHTEIVKALNAFCKEKGILSGSINGIGAIGELTLRFFNPKTKAYDDKTFREQMEISNLTGNISSMNEQVYLHLHITVGRSDYSALAGHLLSAIQNGAGEFVVEDYSERISRTYNPDLGLNIYDFER
>T=0.1, sample=1, score=0.7291, global_score=0.9330, seq_recovery=0.5736
NMYSYKKIGNKYIVSINNHTEIVKALKKFCEEKNIKSGSVNGIGSIGSVTLKFYNLETKEEELKTFNANFEISNLTGFISMHDNKVFLDLHITIGDENFSALAGHLVSAVVNGTCELIVEDFNELVSTKYNEELGLWLLDFEK/NMYSYKKIGNKYIVSINNHTDIVTAIKKFCEDKKIKSGTINGIGQVKEVTLEFRNFETGEKEEKTFKKQFTISNLTGFISTKDGKVFLDLHITFGDENFSALAGHLISAIVDGKCELIIEDYNEEINVKYNEELGLYLLDFNK
>T=0.1, sample=2, score=0.7414, global_score=0.9355, seq_recovery=0.6075
NMYKYKKIGNKYIVSINNHTEIVKAIKEFCKEKNIKSGTINGIGQVGKVTLRFYNPETKEYTEKTFNDNFEISNLTGFISTYKNEVFLHLHITFGKSDFSALAGHLLSAIVNGICELIVEDFKENLSMKYDEKTGLYLLDFEK/NMYKYKKIGNKYVVSINNHTEIVEALKAFCEDKKIKSGTVNGIGQVSKVTLKFFNIETKESKEKTFNKNFEISNLTGFISEINGEVFLHLHITIGDENFSALAGHLLSAVVNGEAILIVEDYKEKVNRKYNEELGLNLLDFNL
score- average over residues that were designed negative log probability of sampled amino acidsglobal score- average over all residues in all chains negative log probability of sampled/fixed amino acidsfixed_chains- chains that were not designed (fixed)designed_chains- chains that were redesignedmodel_name/CA_model_name- model name that was used to generate results, e.g.v_48_020git_hash- github version that was used to generate outputsseed- random seedT=0.1- temperature equal to 0.1 was used to sample sequencessample- sequence sample number 1, 2, 3...etc
@article{dauparas2022robust,
title={Robust deep learning--based protein sequence design using ProteinMPNN},
author={Dauparas, Justas and Anishchenko, Ivan and Bennett, Nathaniel and Bai, Hua and Ragotte, Robert J and Milles, Lukas F and Wicky, Basile IM and Courbet, Alexis and de Haas, Rob J and Bethel, Neville and others},
journal={Science},
volume={378},
number={6615},
pages={49--56},
year={2022},
publisher={American Association for the Advancement of Science}
}
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed

Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file proteinmpnn_mps-1.0.2.tar.gz.
File metadata
- Download URL: proteinmpnn_mps-1.0.2.tar.gz
- Upload date:
- Size: 68.9 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1ef2c9749e869020a802b363e80435ffd3564f5ebe68a7bb86331955b3eb7671
|
|
| MD5 |
4d73041cf2f7e2470e8fc0a7a8da7707
|
|
| BLAKE2b-256 |
aa7bd32f7986f70c1ba2d4d3ecb2b70f5b12946434c9a3bacfb1cb50fa97a1a5
|
Provenance
The following attestation bundles were made for proteinmpnn_mps-1.0.2.tar.gz:
Publisher:
publish.yml on tmsincomb/ProteinMPNN
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
proteinmpnn_mps-1.0.2.tar.gz -
Subject digest:
1ef2c9749e869020a802b363e80435ffd3564f5ebe68a7bb86331955b3eb7671 - Sigstore transparency entry: 1421305148
- Sigstore integration time:
-
Permalink:
tmsincomb/ProteinMPNN@1e11df004ea089bf9323331700b56acc280c3855 -
Branch / Tag:
refs/tags/v1.0.2 - Owner: https://github.com/tmsincomb
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@1e11df004ea089bf9323331700b56acc280c3855 -
Trigger Event:
push
-
Statement type:
File details
Details for the file proteinmpnn_mps-1.0.2-py3-none-any.whl.
File metadata
- Download URL: proteinmpnn_mps-1.0.2-py3-none-any.whl
- Upload date:
- Size: 68.1 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
01af9b4d32e923c2d114931352431cce5598da7efbc6983fcc0a10b7ccc95209
|
|
| MD5 |
004fb9cf2d06c006ce76d9c9c9cad209
|
|
| BLAKE2b-256 |
2594a4c87f6d7bab4c57627995bde0ba5662a0fe9401e1a4cb3abddc6b490d78
|
Provenance
The following attestation bundles were made for proteinmpnn_mps-1.0.2-py3-none-any.whl:
Publisher:
publish.yml on tmsincomb/ProteinMPNN
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
proteinmpnn_mps-1.0.2-py3-none-any.whl -
Subject digest:
01af9b4d32e923c2d114931352431cce5598da7efbc6983fcc0a10b7ccc95209 - Sigstore transparency entry: 1421305279
- Sigstore integration time:
-
Permalink:
tmsincomb/ProteinMPNN@1e11df004ea089bf9323331700b56acc280c3855 -
Branch / Tag:
refs/tags/v1.0.2 - Owner: https://github.com/tmsincomb
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@1e11df004ea089bf9323331700b56acc280c3855 -
Trigger Event:
push
-
Statement type:







