A Python package for extracting protein sequence features

Project description

A Python package for extracting protein sequence features
Documentation
·
Request a feature
·
Report a bug







protlearn is a Python package for the feature extraction of amino acid sequences. It is comprised of three stages - preprocessing, feature computation, and subsequent dimensionality reduction. Currently, the package is being maintained for Python versions 3.6, 3.7, and 3.8.
Overview
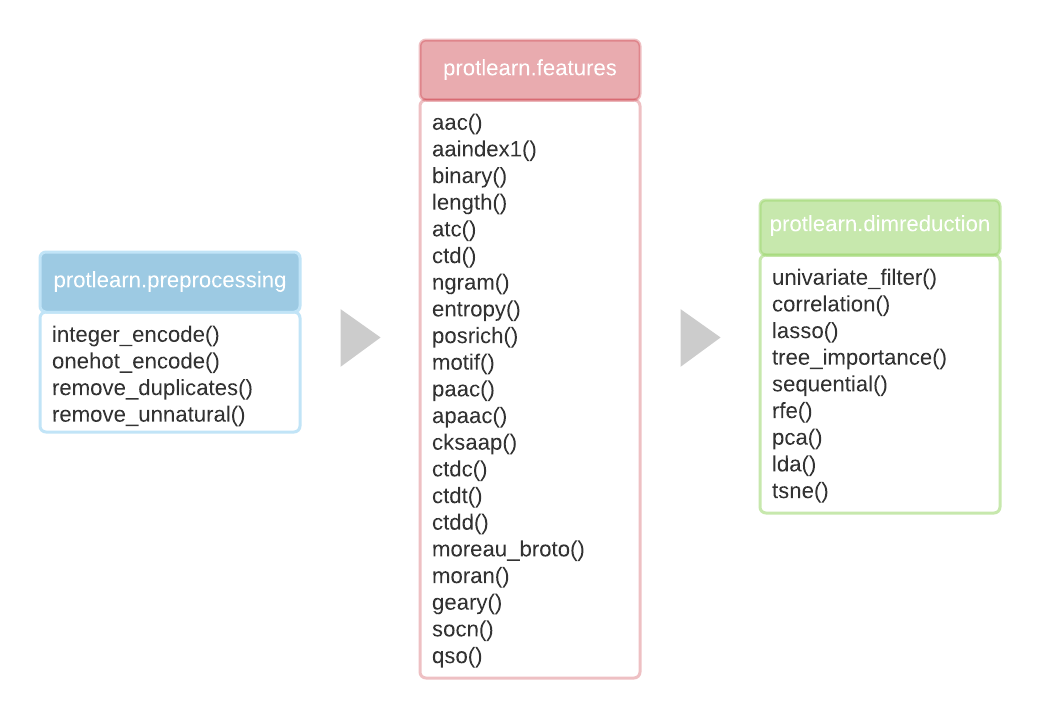
For more information on how to use it, please refer to the documentation.
Installation
Dependencies
- NumPy
- Pandas
- scikit-learn
- xgboost
- mlxtend
- biopython
User Installation
PyPI
You can install protlearn with pip:
$ pip install protlearn
Conda
You can install protlearn with conda:
$ conda install -c conda-forge protlearn
Authors
This package is maintained by Thomas Dorfer.
License
This package is licensed under the MIT License.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file protlearn-0.0.3.tar.gz.
File metadata
- Download URL: protlearn-0.0.3.tar.gz
- Upload date:
- Size: 67.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.2.0 pkginfo/1.5.0.1 requests/2.24.0 setuptools/49.2.0.post20200714 requests-toolbelt/0.9.1 tqdm/4.47.0 CPython/3.8.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
093fecebecd98da885b6d76588d510b42c15dfa65384ac52ebdc95d80ce4f1f8
|
|
| MD5 |
7b2dc9e1e17190e7d2f2d98f337c1f76
|
|
| BLAKE2b-256 |
8dc14ae63ac9d1cd0ec1b98cda53998ca9d33236b876a1c9c9659c3d34e5cad0
|
File details
Details for the file protlearn-0.0.3-py3-none-any.whl.
File metadata
- Download URL: protlearn-0.0.3-py3-none-any.whl
- Upload date:
- Size: 106.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.2.0 pkginfo/1.5.0.1 requests/2.24.0 setuptools/49.2.0.post20200714 requests-toolbelt/0.9.1 tqdm/4.47.0 CPython/3.8.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5288870261df1b3b0072a1997004d83a25d197db4f2589a0b219baeddde1cfb8
|
|
| MD5 |
a79efdc883c8b70f28c0f487cf884e8e
|
|
| BLAKE2b-256 |
881f1c85f75779664f65bdaac78a515bcf86ea0c8926c74fa356c138b447dfbc
|







