A Python PiPeLine framework

Project description
PyPPL - A Python PiPeLine framework







Documentation | API | Change log
Features
- Process caching.
- Process reusability.
- Process error handling.
- Runner customization.
- Easy running profile switching.
- Plugin system.
Installation
pip install PyPPL
Plugin gallery
(*) shipped with PyPPL
- pyppl_report: Generating reports for PyPPL pipelines
- pyppl_flowchart: Generating flowchart for PyPPL
- pyppl_export*: Exporting outputs generated by PyPPL pipeline
- pyppl_echo*: Echoing script output to PyPPL logs"
- pyppl_rich*: Richer information in logs for PyPPL
- pyppl_strict*: More strict check of job success for PyPPL
- pyppl_lock*: Preventing running processes from running again for PyPPL
- pyppl_annotate: Adding long description/annotation for processes
- pyppl_require: Checking and installing requirements for processes
- pyppl_jobtime: Job running time statistics for PyPPL
- pyppl_notify: Email notifications for PyPPL
- pyppl_runcmd: Allowing to run local command before and after each process for PyPPL
- pyppl_runners: Common runners for PyPPL
Writing pipelines with predefined processes
Let's say we are implementing the TCGA DNA-Seq Re-alignment Workflow (The very left part of following figure). For demonstration, we will skip the QC and the co-clean parts here.
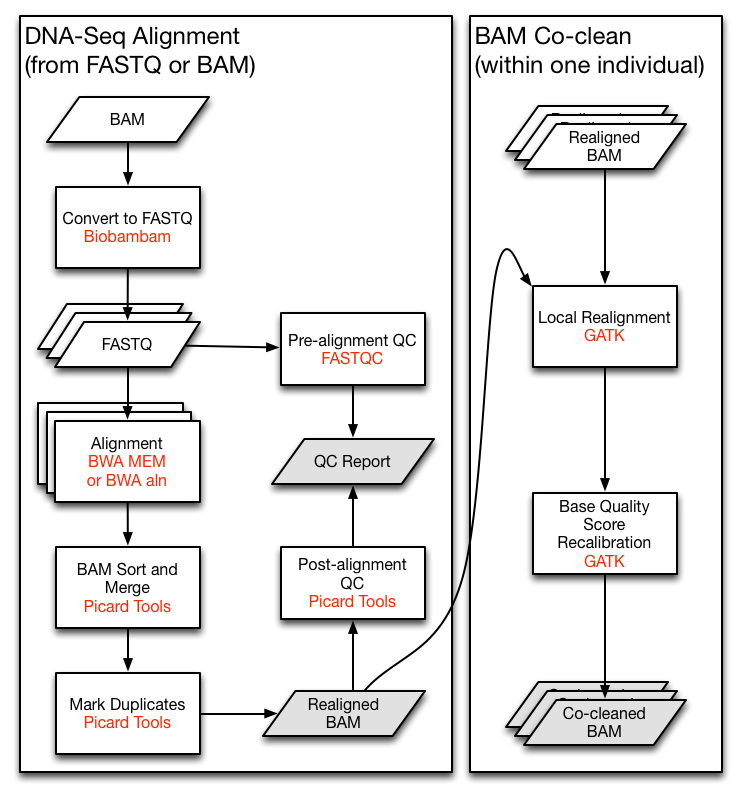
demo.py:
from pyppl import PyPPL, Channel
# import predefined processes
from TCGAprocs import pBamToFastq, pAlignment, pBamSort, pBamMerge, pMarkDups
# Load the bam files
pBamToFastq.input = Channel.fromPattern('/path/to/*.bam')
# Align the reads to reference genome
pAlignment.depends = pBamToFastq
# Sort bam files
pBamSort.depends = pAlignment
# Merge bam files
pBamMerge.depends = pBamSort
# Mark duplicates
pMarkDups.depends = pBamMerge
# Export the results
pMarkDups.config.export_dir = '/path/to/realigned_Bams'
# Specify the start process and run the pipeline
PyPPL().start(pBamToFastq).run()

Implementing individual processes
TCGAprocs.py:
from pyppl import Proc
pBamToFastq = Proc(desc = 'Convert bam files to fastq files.')
pBamToFastq.input = 'infile:file'
pBamToFastq.output = [
'fq1:file:{{i.infile | stem}}_1.fq.gz',
'fq2:file:{{i.infile | stem}}_2.fq.gz']
pBamToFastq.script = '''
bamtofastq collate=1 exclude=QCFAIL,SECONDARY,SUPPLEMENTARY \
filename= {{i.infile}} gz=1 inputformat=bam level=5 \
outputdir= {{job.outdir}} outputperreadgroup=1 tryoq=1 \
outputperreadgroupsuffixF=_1.fq.gz \
outputperreadgroupsuffixF2=_2.fq.gz \
outputperreadgroupsuffixO=_o1.fq.gz \
outputperreadgroupsuffixO2=_o2.fq.gz \
outputperreadgroupsuffixS=_s.fq.gz
'''
pAlignment = Proc(desc = 'Align reads to reference genome.')
pAlignment.input = 'fq1:file, fq2:file'
# name_1.fq.gz => name.bam
pAlignment.output = 'bam:file:{{i.fq1 | stem | stem | [:-2]}}.bam'
pAlignment.script = '''
bwa mem -t 8 -T 0 -R <read_group> <reference> {{i.fq1}} {{i.fq2}} | \
samtools view -Shb -o {{o.bam}} -
'''
pBamSort = Proc(desc = 'Sort bam files.')
pBamSort.input = 'inbam:file'
pBamSort.output = 'outbam:file:{{i.inbam | basename}}'
pBamSort.script = '''
java -jar picard.jar SortSam CREATE_INDEX=true INPUT={{i.inbam}} \
OUTPUT={{o.outbam}} SORT_ORDER=coordinate VALIDATION_STRINGENCY=STRICT
'''
pBamMerge = Proc(desc = 'Merge bam files.')
pBamMerge.input = 'inbam:file'
pBamMerge.output = 'outbam:file:{{i.inbam | basename}}'
pBamMerge.script = '''
java -jar picard.jar MergeSamFiles ASSUME_SORTED=false CREATE_INDEX=true \
INPUT={{i.inbam}} MERGE_SEQUENCE_DICTIONARIES=false OUTPUT={{o.outbam}} \
SORT_ORDER=coordinate USE_THREADING=true VALIDATION_STRINGENCY=STRICT
'''
pMarkDups = Proc(desc = 'Mark duplicates.')
pMarkDups.input = 'inbam:file'
pMarkDups.output = 'outbam:file:{{i.inbam | basename}}'
pMarkDups.script = '''
java -jar picard.jar MarkDuplicates CREATE_INDEX=true INPUT={{i.inbam}} \
OUTPUT={{o.outbam}} VALIDATION_STRINGENCY=STRICT
'''
Each process is indenpendent so that you may also reuse the processes in other pipelines.
Pipeline flowchart
# When try to run your pipline, instead of:
# PyPPL().start(pBamToFastq).run()
# do:
PyPPL().start(pBamToFastq).flowchart().run()
Then an SVG file endswith .pyppl.svg will be generated under current directory.
Note that this function requires Graphviz and graphviz for python.
See plugin details.

Pipeline report
See plugin details
pPyClone.report = """
## {{title}}
PyClone[1] is a tool using Probabilistic model for inferring clonal population structure from deep NGS sequencing.
}})
```table
caption: Clusters
file: "{{path.join(job.o.outdir, "tables/cluster.tsv")}}"
rows: 10
```
[1]: Roth, Andrew, et al. "PyClone: statistical inference of clonal population structure in cancer." Nature methods 11.4 (2014): 396.
"""
# or use a template file
pPyClone.report = "file:/path/to/template.md"
PyPPL().start(pPyClone).run().report('/path/to/report', title = 'Clonality analysis using PyClone')

Full documentation
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file PyPPL-3.2.2.tar.gz.
File metadata
- Download URL: PyPPL-3.2.2.tar.gz
- Upload date:
- Size: 62.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.0.5 CPython/3.7.3 Linux/3.10.0-1062.12.1.el7.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8a37c56f0aeed8aa72028a6ff29689edd299021427f7e9e87bdae6984b9d6485
|
|
| MD5 |
f77e77c4b17941472dac5be30797fffa
|
|
| BLAKE2b-256 |
31caef68bb56d23cdf3e961bf1f3edcc49c83f7ea098fff25380aeabdd0bf912
|
File details
Details for the file PyPPL-3.2.2-py3-none-any.whl.
File metadata
- Download URL: PyPPL-3.2.2-py3-none-any.whl
- Upload date:
- Size: 67.7 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.0.5 CPython/3.7.3 Linux/3.10.0-1062.12.1.el7.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
40a2a79c6dfcfd5848520e9909723c3b1f3a534ac047c55554dde2b920ee8787
|
|
| MD5 |
41cbc2e07346a912187e1185fe2c603c
|
|
| BLAKE2b-256 |
220fc5475ddc0bc5d6e959a4182c875a1864204cf4883960456dc8b34f43cf7e
|







