Deconvolve CUT&Tag 2for1 data.

Project description
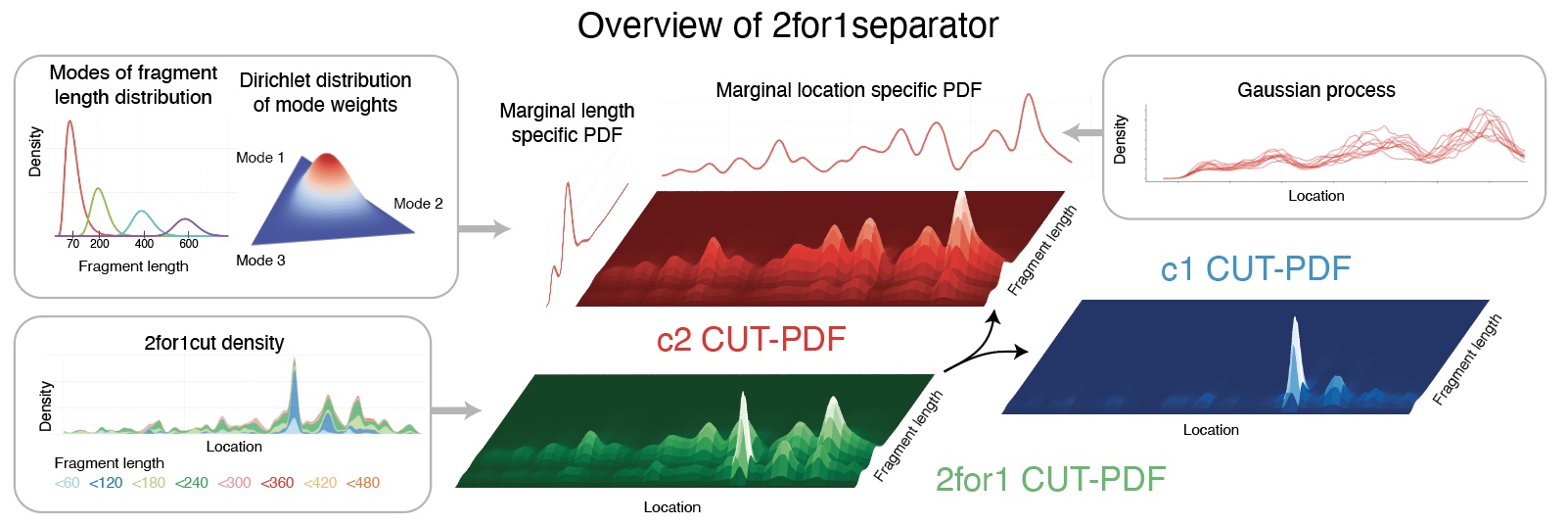
2for1separator

2for1 separator is an algorithm to deconvolve CUT&Tag2for1 data. It uses differences in the fragment length distributions of the two targets and the proximity of chromatin cuts to estimate the probability for each cut to originate from one target or the other. The result is a set of cut density tracks that represent the estimated number of cuts induced by the two antibodies used in the CUT&Tag2for1 experiment.
Installation
Using conda
TBA
Using pip
Please make sure python points to a Python 3.9+ interpreter
and libcurl is installed.
We highly recommed to install install
scikit-sparse
to significantly reduce memory demand and runtime.
Finally, inslatt 2for1separator with
pip install sep241
From source
To install from source you can run:
git clone https://github.com/settylab/2for1separator
cd 2for1separator
pip install .
Usage
Before the deconvolution, the data has to be split up into manageable chunks:
sep241prep [bed files] --out [jobdata pkl file] --memory [max memory target in GB]
It is recommended to use approximately 20 GB or more for --memory.
This specifies the targeted memory demand during the subsequent deconvolution.
The output of the function reports the number of separate work chunks
and suggests subsequent calls for the deconvolution.
The number of slurm jobs is also stored in an additional output file named
with [jobdata pkl file].njobs.
Note that if memory resources are exhausted, deconvolution jobs
may be cancled by slurm or the operating system and subsequent
results will be missing. The downstream scripts will look for missing
results and report a comma sperated list of respective work chunk
numbers. If you are using slurm you can rerun the slurm jobs of only
the specified jobs by passing the list with the --array= parameter
of the sbatch command.
Exporting Results
If not specified otherwise through the --out argument, all outputs are placed
into the same directory with the [jobdata pkl file]. Most outputs contain
c1 or c2 in their file name, which stand for channel 1 or channel 2,
and represent the two constituent parts of the data that were
induced by the two different targets and that were
reconstructed through the deconvolution.
To produce bigwig files from the deconvolution results run
sep241mkbw [jobdata pkl file] [chrom sizes file]
The chomosome sizes file needs to have two columns with chromosome name and
size in bases
(see bigWIG format).
The produced bigWIG files may be used for downstream analysis such as peak calling. To use the 2for1seperator cut-likelihood-based peak calling with overlap identification run:
sep241peakcalling [jobdata pkl file]
Note, that sep241peakcalling does not require the prior conversion
to bigWIG but instead uses the raw deconvolution output.
To write out the target specific likelihoods of each genomic cut you can run
sep241events [jobdata pkl file]
For more information pass --help to the respective commands.
Visualization
Output files are bigwigs and bed-files. These can be visualised with software tools such as the IGV Browser or JBrowser 2. Visualization of intermediate results are currently only possible if intermediate function calls within the supplied scripts are reproduced in a python environment.
Citation
Janssens, D.H., Otto, D.J., Meers, M.P. et al. CUT&Tag2for1: a modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol 23, 81 (2022). https://doi.org/10.1186/s13059-022-02642-w


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file sep241-0.4.5.tar.gz.
File metadata
- Download URL: sep241-0.4.5.tar.gz
- Upload date:
- Size: 58.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.0 CPython/3.9.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
21bb02b76cdeef3c57baaaa1cd0fa70993fd5c7f48802a28ae47865a23ec069c
|
|
| MD5 |
566124d1656bd3bde180f30ffd44bc2c
|
|
| BLAKE2b-256 |
a93416c68b52a22c614d121af105a71a4ab10e6e3fd3004a6644f7de1e072301
|







