Run assembler (Canu, Flye, Hifiasm) on a set of long read files

Project description




LORA — Long Read Assembly pipeline
- Overview:
Assemble long reads (PacBio HiFi, PacBio subreads, Nanopore) into high-quality genome assemblies with optional polishing, annotation, and quality assessment.
- Input:
BAM files from PacBio sequencers, or FastQ files from Nanopore or PacBio HiFi sequencers.
- Output:
HTML reports with per-sample assembly statistics, coverage, BLAST identification, BUSCO scores, and optional annotation.
- Status:
Production
- Citation:
- Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines,
Journal of Open Source Software, 2(16), 352, doi:10.21105/joss.00352
- Zenodo DOI:
-
- Biorxiv:
https://www.biorxiv.org/content/10.64898/2026.01.06.697901v1

Installation
pip install sequana-lora
To upgrade an existing installation:
pip install sequana-lora --upgrade
Quick Start
Step 1 — prepare the working directory:
sequana_lora \
--input-directory /path/to/reads \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /path/to/containers
This creates a lora/ working directory containing config.yaml and a lora.sh launch script.
Step 2 — review the configuration (optional but recommended):
cd lora cat config.yaml # adjust parameters as needed
Step 3 — run the pipeline:
sh lora.sh
Or launch directly from step 1 with --execute (skips the review step):
sequana_lora ... --execute
To watch live progress in the terminal, add --monitor:
sequana_lora ... --execute --monitor
Required options
Three options are always required:
- --assembler
Assembler to use. Choices: flye (recommended for HiFi), canu, hifiasm, unicycler, necat, pecat.
- --data-type
Technology and quality of the input reads:
Value
Description
pacbio-hifi
PacBio HiFi / CCS reads (Q20+)
pacbio-raw
PacBio CLR / subreads (raw)
pacbio-corr
PacBio corrected reads
nano-hq
Nanopore Q20+ reads (e.g. R10.4 with SUP basecalling)
nano-raw
Nanopore standard reads
nano-corr
Nanopore corrected reads
- --genome-size
Estimated genome size, e.g. 3m (3 Mb), 2.5g (2.5 Gb). Required by Flye; used by Canu for coverage reporting.
Common Examples
PacBio HiFi (recommended setup)
sequana_lora \
--input-directory /data/hifi \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /shared/containers \
--do-coverage
Nanopore (bacteria, full quality pipeline)
Use --mode bacteria to enable sequana_coverage, prokka, busco, and checkm in one shot:
sequana_lora \
--input-directory /data/nanopore \
--data-type nano-hq \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /shared/containers \
--mode bacteria \
--busco-lineage bacteria \
--checkm-rank genus \
--checkm-name Streptococcus
PacBio subreads (with CCS construction)
If your input is raw PacBio BAM files (subreads), LORA can build CCS/HiFi reads first:
sequana_lora \
--input-directory /data/subreads \
--data-type pacbio-raw \
--assembler flye \
--genome-size 3m \
--pacbio-build-ccs \
--pacbio-ccs-min-passes 10 \
--pacbio-ccs-min-rq 0.99
Or, if you have multiple BAM files per sample, provide a CSV:
sequana_lora \
--pacbio-input-csv samples.csv \
--data-type pacbio-raw \
--assembler flye \
--genome-size 3m
The CSV format is: one row per sample with columns sample,file1[,file2,...].
Optional Steps
Coverage analysis
Computes depth of coverage and breadth of coverage for each contig using sequana_coverage. Highly recommended to check assembly quality:
--do-coverage
BUSCO completeness
Assess genome completeness against a lineage-specific marker gene set:
--busco-lineage bacteria # auto-download bacteria lineage --busco-lineage streptococcales # specific clade --busco-print-lineages # list all available lineages
Prokka annotation
Annotate contigs (bacterial genomes):
--do-prokka
CheckM genome quality
Estimate completeness and contamination for bacterial genomes:
--checkm-rank genus --checkm-name Streptococcus
Use an invalid --checkm-name value to get a list of valid names for a given rank, e.g. --checkm-rank genus --checkm-name HELP.
Polypolish (Illumina polishing)
Polish long-read contigs with paired-end Illumina data:
--do-polypolish \ --polypolish-input-directory /data/illumina \ --polypolish-input-pattern "*.fastq.gz" \ --polypolish-input-readtag "_R[12]_"
Circularisation
Explicit circularisation with Circlator (Flye performs this automatically):
--do-circlator
BLAST identification
BLAST aligns each contig against a nucleotide database to identify the assembled sequences. The top hits appear in the HTML report.
Local BLAST
Requires a locally installed BLAST+ and a downloaded nt database (~270 GB). Fastest option with no network dependency:
--blastdb /path/to/blast/databases
Remote BLAST (NCBI)
No local database required — jobs are submitted to NCBI’s BLAST servers. Enable by providing an email address:
--blast-email your@email.com
Jobs are submitted sequentially (one contig at a time) to avoid IP-level CPU throttling by NCBI. The default database is nt; use --blast-remote-db to change it:
--blast-email your@email.com --blast-remote-db refseq_genomic
Restricting the search to an organism group (strongly recommended)
Searching all of nt for a large contig is slow and prone to NCBI CPU throttling. Restrict the search by editing config.yaml after running sequana_lora. The entrez_query parameter is equivalent to filling the “Organism” box on the NCBI BLAST web form.
Option 1 — curated bacterial reference genomes (fastest, recommended for bacteria)
Use refseq_genomic as the database and restrict to bacteria. RefSeq contains only complete, curated reference genomes — a much smaller and higher-quality search space than nt:
blast:
remote_db: 'refseq_genomic'
entrez_query: 'Bacteria[Organism]'
Option 2 — all bacteria in nt, reference sequences only
Stay on nt but filter to RefSeq-quality entries:
blast:
remote_db: 'nt'
entrez_query: 'Bacteria[Organism] AND refseq[filter]'
Option 3 — single genus
Useful when the organism is known:
blast:
remote_db: 'nt'
entrez_query: 'Streptococcus[Organism]'
NCBI API key (optional but recommended)
Register for a free NCBI API key at https://www.ncbi.nlm.nih.gov/account/ (sign in → API Key Management). It raises the rate limit from 3 to 10 requests/second and reduces CPU throttling for large queries. Add it to config.yaml:
blast:
api_key: 'YOUR_KEY_HERE'
HPC / SLURM cluster
On a cluster with SLURM, pass --profile slurm:
sequana_lora \
--input-directory /data/hifi \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--profile slurm \
--slurm-queue fast \
--jobs 40 \
--apptainer-prefix /shared/containers
Per-rule memory and thread settings are controlled via the resources blocks in config.yaml.
Apptainer / Singularity (no system installs needed)
Every tool runs inside a pre-built container. Point --apptainer-prefix to a shared directory so images are downloaded once and reused across projects:
--apptainer-prefix /shared/containers
Images are downloaded automatically on first run from Zenodo. Pass extra bind mounts with --apptainer-args if your data lives outside $HOME:
--apptainer-args "-B /data:/data"
Configuration file
After running sequana_lora, a config.yaml is created in the working directory. All pipeline parameters can be tuned there. Key sections:
assembler — which assembler to use
flye / canu / hifiasm — assembler-specific options
fastp — read filtering (minimum length, etc.)
blast — BLAST settings including entrez_query and api_key
busco / prokka / checkm — optional QC tools
sequana_coverage — coverage analysis parameters
multiqc — aggregated report settings
Full reference: config.yaml
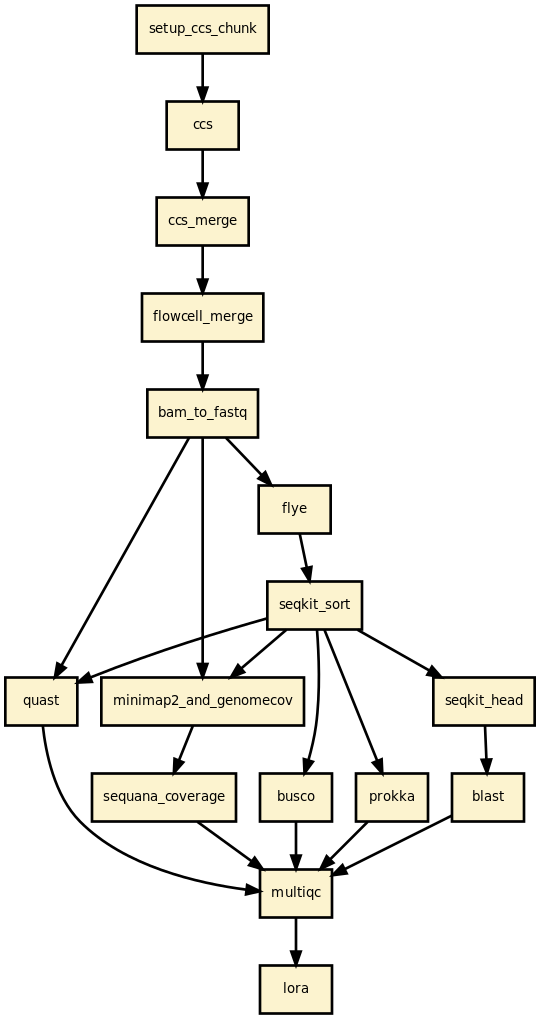
Pipeline overview
Read filtering — fastp removes reads below the minimum length threshold.
[Optional] CCS — build HiFi reads from PacBio subreads (ccs tool).
Assembly — Flye / Canu / Hifiasm / Unicycler / NECAT / PECAT.
[Optional] Circularisation — Circlator (or built into Flye).
[Optional] Polishing — Polypolish with paired-end Illumina reads.
Contig sorting — SeqKit sorts contigs by length (largest first).
Read mapping — Minimap2 maps reads back to contigs; Mosdepth computes coverage.
[Optional] Coverage analysis — sequana_coverage per contig.
Quality assessment — QUAST assembly statistics.
[Optional] BLAST — top hits per contig (local or remote NCBI).
[Optional] BUSCO — genome completeness.
[Optional] Prokka — genome annotation.
[Optional] CheckM — contamination and completeness for bacteria.
Reports — per-sample HTML report and a multi-sample summary.
Changelog
Version |
Description |
|---|---|
1.1.0 |
|
1.0.0 |
|
0.3.0 |
|
0.2.0 |
|
0.1.0 |
First release. |
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sequana_lora-1.1.0.tar.gz.
File metadata
- Download URL: sequana_lora-1.1.0.tar.gz
- Upload date:
- Size: 192.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/2.0.1 CPython/3.10.14 Linux/6.14.5-100.fc40.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e8ce39b680195bfd3ef4b779e59313f11a1c4fc422c767ec95833cda8cdd8c8d
|
|
| MD5 |
a68d55df35a5b96f9ca59846eae13d02
|
|
| BLAKE2b-256 |
b7569fa65e2a82a924f701f25cc4fc99dbc4b3f79d1929420d6b63c551389b5b
|
File details
Details for the file sequana_lora-1.1.0-py3-none-any.whl.
File metadata
- Download URL: sequana_lora-1.1.0-py3-none-any.whl
- Upload date:
- Size: 192.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/2.0.1 CPython/3.10.14 Linux/6.14.5-100.fc40.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a105eb8f2491a5a6115570554c1b3f8d438253a4be74ec09c747a7d89fd0d492
|
|
| MD5 |
317df9d8ae8f1f635e64a1d28e435d71
|
|
| BLAKE2b-256 |
311688ad486f1347bdb99220dfd9e053c13d39d746e7138f7f48a6f153e8ce5d
|







