Synthetic data generator for snail mutation survey

Project description
Snailz

These data generators model genomic analysis of snails in the Pacific Northwest that are growing to unusual size as a result of exposure to pollution.
- One or more surveys are conducted at one or more sites.
- Each survey collects genomes and sizes of snails.
- A grid at each site is marked out to show the presence or absence of pollution.
- Laboratory staff perform assays of the snails' genetic material.
- Each assay plate has a design showing the material applied and readings showing the measured response.
- Plates may be invalidated after the fact if a staff member believes it is contaminated.
Usage
- Create a fresh Python environment:
uv venv - Activate that environment:
source .venv/bin/activate - Build development version of package:
uv pip install -e . - View available commands:
snailz --help - Copy default parameter files:
snailz params --outdir ./params - See how to regenerate datasets:
python -c 'import snailz; help(snailz)'
To regenerate all data using the default parameters provided, run:
snailz everything --paramsdir ./params --datadir ./data --verbose
Database
The final database data/lab.db is structured as shown below.
Note that the data from the file assays.json is split between several tables.
Note also that the SQLite database file is not included in this repository
because its binary representation changes each time it is regenerated
(even though the values it contains stay the same).
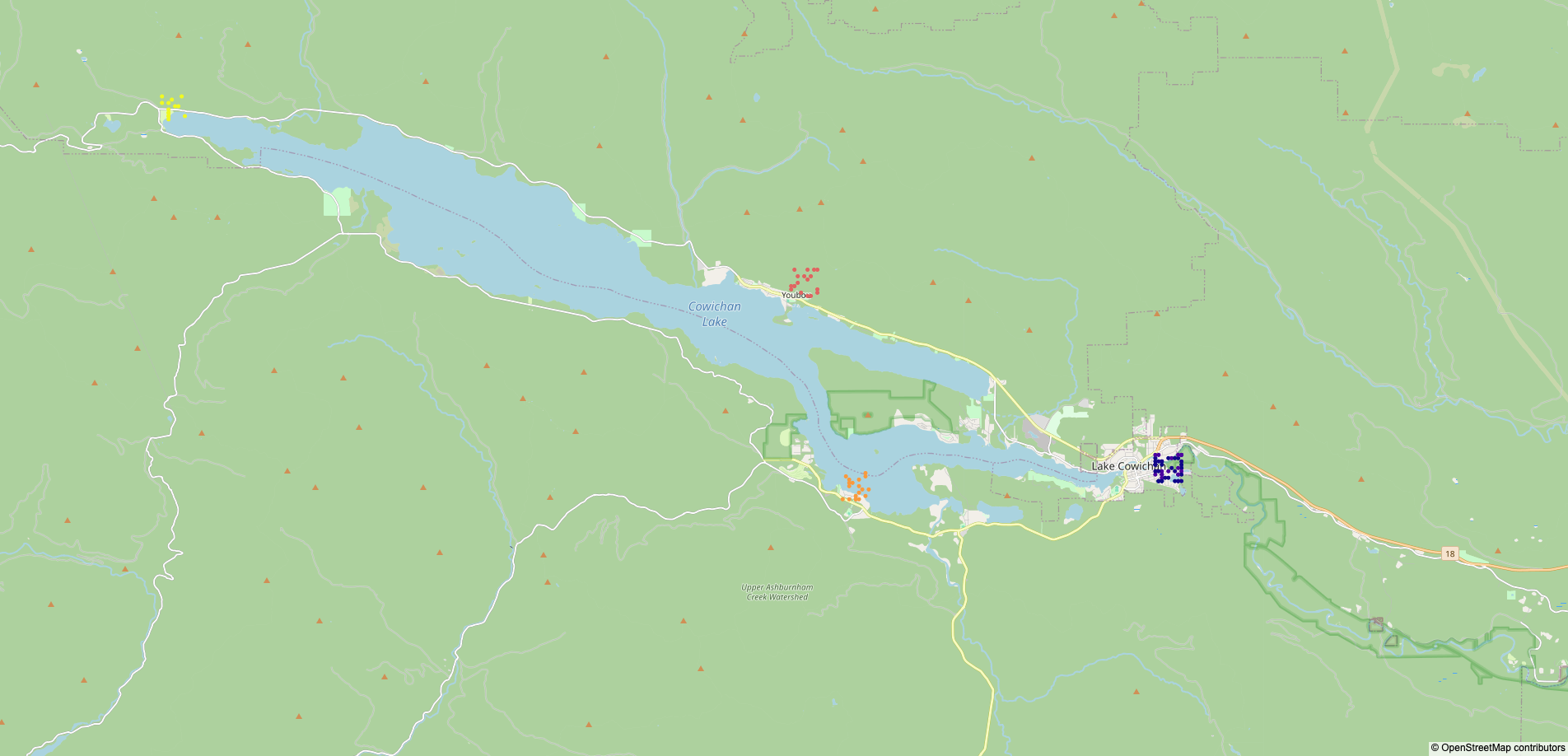
The map of survey locations in data/survey.png is not included in the repository for the same reason,
but a duplicate is manually saved in img/survey.png.

site: survey sitesite_id: primary key (text)lon: longitude of site reference marker (float deg)lat: latitude of site reference marker (float deg)
surveysurvey_id: primary key (text)site_id: foreign key of site where survey was conducted (text)date: date that survey was conducted (date, YYYY-MM-DD)
sample: sample taken from surveysample_id: primary key (int, 1-1 withexperiment.sample_id)survey_id: foreign key of survey (int)lon: longitude of sample site (float deg)lat: latitude of sample site (float deg)sequence: genome sequence of sample (text)size: snail size (float)
experiment: experiment done on samplesample_id: primary key (int, 1-1 withsample.sample_id)kind: kind of experiment (text, either 'ELISA' or 'JESS')start: start date (date, YYYY-MM-DD)end: end date (date, YYYY-MM-DD, null if experiment is ongoing)
staffstaff_id: primary key (int)personal: personal name (text)family: family name (text)
performed: join table showing which staff members performed which experimentsstaff_id: foreign key of staff membersample_id: foreign key of sample/experiment
plate: information about single assay plateplate_id: primary key (int)sample_id: foreign key of sample/experiment (int)date: date that plate was run (date, YYYY-MM-DD)filename: filename of design/results file (text)
invalidated: invalidated platesplate_id: foreign key of plate (int)staff_id: foreign key of staff member who did invalidation (int)date: when plate was invalidated
Data Files
./data contains a generated dataset for reference.
As noted above,
it does not contain the SQLite database file lab.db;
run snailz db to regenerate it.
(See help(snailz) for an example invocation.)
- Staff:
staff.csvstaff_id: unique staff member identifier (int > 0)personal: personal name (text)family: family name (text)
- Genomes:
genomes.jsonlength: number of base pairs (int > 0)reference: the unmutated reference genome (text)individuals: sequences for individuals (list of text)locations: locations of mutations (list of int)susceptible_loc: location of mutation of interest (int >= 0)susceptible_base: mutated base responsible for size change (char)
- Grids:
grids/*.csv(one file per site)- values are contamination levels at sample points (0 means no contamination)
- Samples:
grids/samples.csvsample_id: unique ID for genetic sample (text)survey_id: which survey it was taken in (text)lon: longitude of sample site (float)lat: latitude of sample site (float)sequence: sampled gene sequence (text)size: snail weight (float, grams)
- Assays:
assays.jsonexperiment: experiment detailssample_id: sample that experiment used (int > 0)kind: "ELISA" or "JESS" (text)start: start date (date, YYYY-MM-DD)end: end date (date, YYYY-MM-DD or None if experiment incomplete)
performed: join table showing who performed which experimentsstaff_id: foreign key tostaffsample_id: foreign key toexperiment
plate: details of assay plates used in experimentsplate_id: unique plate identifier (int > 0)sample_id: foreign key tosample(text)date: date plate was run (date, YYYY-MM-DD)filename: name of design and results files (text)
invalidated: which plates have been invalidatedplate_id: foreign key to plate (text)staff_id: foreign key to staff member responsible (text)date: invalidation date (date, YYYY-MM-DD)
- Plates are represented by matching files in the
designsandreadingsdirectoriesdesigns/*.csv: assay plate designs- header: machine type, file type ("design" or "readings"), staff ID
- blank line
- table with column and row titles showing material in each well
readings/*.csv: assay plate readings- header: machine type, file type ("design" or "readings"), staff ID
- blank line
- table with column and row titles showing reading from each well
- To simulate the messiness of real experimental data,
the tidy assay plate files in
readings/*.csvare copied tomangled/*.csvwith random changes:- Some files have a staff member's name added in the first row.
- Some have an extra header row containing the experiment date.
- Some have a footer with the staff member's ID.
- In some, the values are offset one column to the right.
Workflow
The workflow used to generate the database and data files is shown below:
snailzorsnailz --help: show available commandssnailz clean: remove all datasetssnailz everything: make all datasetssnailz grids: synthesize pollution gridssnailz genomes: synthesize genomessnailz samples: sample snails from survey sitessnailz staff: synthesize staffsnailz assays: generate assay filessnailz plates: generate plate filessnailz mangle: create mangled plate reading filessnailz db: generate databasesnailz map: generate SVG map of sample locations (in progress)

Parameters
./snailz/params contains the parameter files used to control generation of the reference dataset.
These are included in the package and can be copied into the current directory using snailz params --outdir .
(replace . with another directory name as desired).
snailz params also copies a Makefile that can re-run commands with appropriate parameters;
see the table of commands given earlier for options.
- Sites:
sites.csvsite_id: unique label for site (text)lon: longitude of site reference marker (deg)lat: latitude of site reference marker (deg)
- Grids:
grids.jsondepth: range of random values per cell (int > 0)height: number of cells on Y axis (int > 0)seed: RNG seed (int > 0)width: number of cells on X axis (int > 0)
- Surveys:
surveys.csvsurvey_id: unique label for survey (text)site_id: ID of site where survey was conducted (text)date: date that survey was conducted (date, YYYY-MM-DD)spacing: spacing of measurement point (float, meters)
- Genomes:
genomes.jsonlength: number of base pairs in sequences (int > 0)num_genomes: how many individuals to generate (int > 0)num_snp: number of single nucleotide polymorphisms (int > 0)prob_other: probability of non-significant mutations (float in 0..1)seed: RNG seed (int > 0)snp_probs: probability of selecting various bases (list of 4 float summing to 1.0)
- Staff:
staff.jsonlocale: locale to use when generating staff names (text)num: number of staff (int > 0)seed: RNG seed (int > 0)
- Assays:
assays.jsonassay_duration: range of days for each assay (ordered pair of int >= 0)assay_plates: range of plates per assay (ordered pair of int >= 1)assay_staff: range of staff in each assay (ordered pair of int > 0)assay_types: types of assays (list of text)control_val: nominal reading value for control wells (float > 0)controls: labels to used for control wells (list of text)enddate: end of all experimentsfilename_length: length of stem of design/readings filenames (int > 0)fraction: fraction of samples that have been used in experimentsinvalid: probability of plate being invalidated (float in 0..1)seed: RNG seed (int > 0)startdate: start of all experimentsstdev: standard deviation on readings (float > 0)treated_val: nominal reading value for treated well (float > 0)treatment: label to use for treated wells (text)
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file snailz-0.1.16.tar.gz.
File metadata
- Download URL: snailz-0.1.16.tar.gz
- Upload date:
- Size: 25.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
fdce1b499446565bdbb0aa666bc80f0c57a6dda73ef8f536e04bfc582ff8c1e0
|
|
| MD5 |
9a82a4015d57790f2294bfb5d295ea59
|
|
| BLAKE2b-256 |
8d623980df0c2fc703fe9c0db39f5c83392e4deaed2e3117502b343ab83d9b70
|
File details
Details for the file snailz-0.1.16-py3-none-any.whl.
File metadata
- Download URL: snailz-0.1.16-py3-none-any.whl
- Upload date:
- Size: 26.0 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b955712633442e13466e2246fec75830caabe2d548cd3e3e17124ed0ad79b5e9
|
|
| MD5 |
92158eac7e797ae88657ec1be4b53629
|
|
| BLAKE2b-256 |
2517731bac5e659829decaa6a3504f52e6176bda7c9e43ae66d5c2997f614ad5
|







