Publication-quality alignment viewer for nucleotide and amino acid sequences
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
tview
Publication-quality alignment viewer for nucleotide and amino acid sequences. A lightweight alternative to samtools tview that produces clean, stable image output.
Supports BAM files (with reference FASTA), pre-aligned FASTA (e.g. MAFFT output), and stacking multiple inputs into a single figure.
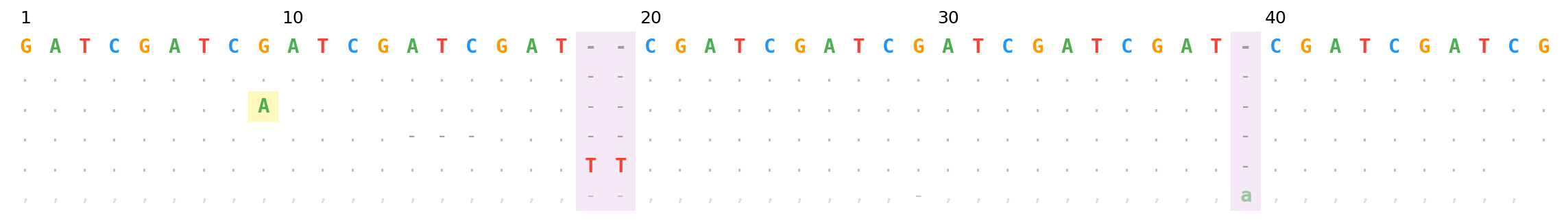


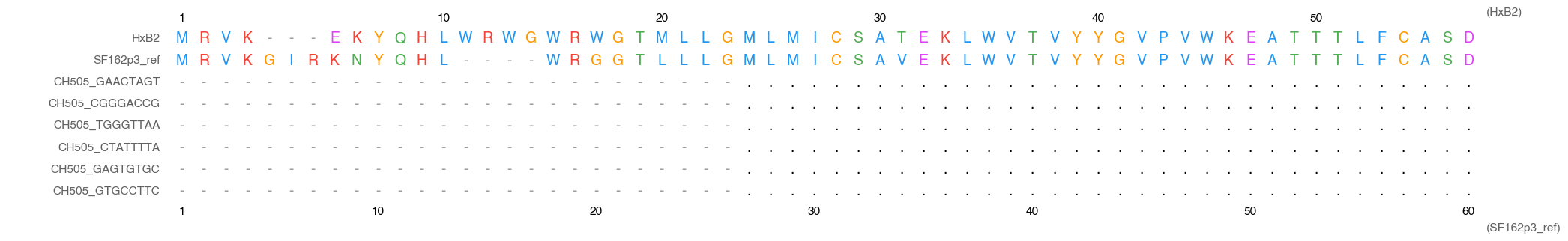
SF162p3_ref (bottom row + bottom x-axis) with HxB2 numbering (top row + top x-axis). Different gap patterns mean position 10 on each axis lands in a different alignment column.
Installation
pip install tview
Installs matplotlib, click, and pysam.
Quick Start
BAM file
tview \
--bam aligned.bam \
--ref reference.fa \
--region chr1:100-200 \
-o alignment.png
Aligned FASTA (e.g. MAFFT output)
The first sequence in the file is treated as the reference.
tview \
--fasta env_protein_aligned.fasta \
--palette aa \
-o env_alignment.png
Subset columns from a FASTA alignment
Use --columns with 1-based inclusive range to window into long alignments.
tview \
--fasta aligned.fasta \
--columns 1-120 \
--palette aa \
-o first_120_cols.png
Classic (black-and-white) mode
Use --classic-mode for textbook-style monochrome output — all black text on a white background with no colored highlighting. Structural conventions (. , lowercase, -) are preserved.
tview \
--fasta aligned.fasta \
--palette aa \
--classic-mode \
-o classic_output.png
Dual Reference (Heterologous MSA)
For heterologous MSAs (e.g. HIV with HxB2 + multiple strains + reads), the variant-call reference and the x-axis numbering reference do not have to be the same sequence. --variant-ref selects the FASTA header against which mismatches are colored; --numbering-ref selects the FASTA header whose non-gap positions label the x-axis.
When both are set and differ:
- Both refs render as rows at the top (numbering ref first, variant ref second).
- A second x-axis is drawn at the bottom of the figure for the variant-ref's coordinates.
- The top axis carries the numbering ref's coordinates with a
(<numbering_ref>)annotation; the bottom axis carries the variant ref's coordinates with a(<variant_ref>)annotation. - Per-row labels are auto-enabled so you can tell the two top rows apart.
--max-rows Ncounts samples only — references are always shown.
tview \
--fasta env_protein_aligned.fasta \
--palette aa \
--columns 1-60 \
--variant-ref SF162p3_ref \
--numbering-ref HxB2 \
--max-rows 6 \
-o env_dual_ref.png
Gap patterns differ between strains — HxB2 position 10 and SF162p3_ref position 10 typically land on different alignment columns. That's the point: both axes complement each other.
Per-row labels
Show sequence IDs to the left of each row (auto-enabled in dual-ref mode):
tview --fasta aligned.fasta --show-row-labels -o labeled.png
Tick frequency
Default labels are placed at non-gap positions 1, 10, 20, 30, ... Use --tick-every N to change the interval. --tick-every 1 labels every column — useful for short alignments or when exact position readout matters.
tview \
--fasta env_protein_aligned.fasta \
--variant-ref SF162p3_ref --numbering-ref HxB2 \
--columns 1-60 --max-rows 6 \
--tick-every 1 \
-o env_dual_ref_per_column.png
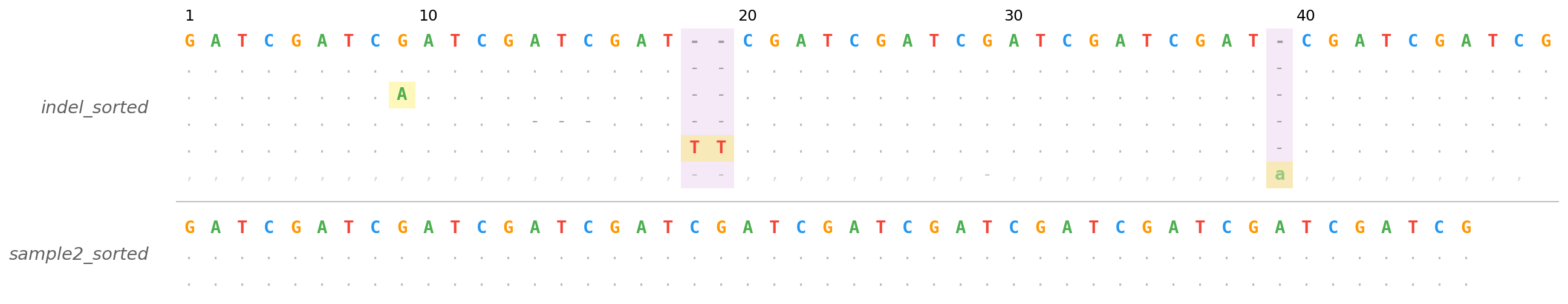
Stacking Multiple Panels
Each input file becomes a vertically stacked panel separated by a thin line. Panels are labeled on the left with the filename stem.
Multiple BAMs (shared reference and region)
tview \
--bam sample1.bam --bam sample2.bam --bam sample3.bam \
--ref reference.fa \
--region chr1:100-200 \
-o stacked.png
Multiple FASTAs
tview \
--fasta group1_aligned.fasta --fasta group2_aligned.fasta \
--palette aa \
--columns 1-120 \
-o comparison.png
Mix BAM and FASTA panels
--ref and --region apply only to BAM panels; --columns applies only to FASTA panels.
tview \
--bam reads.bam \
--ref reference.fa \
--region chr1:100-200 \
--fasta protein_aligned.fasta \
--columns 1-120 \
-o mixed.png
BAM panels are rendered first (top), FASTA panels below.
Piping from stdin
Pass - to read file paths from stdin (one per line). Each path becomes its own panel.
# find → stacked panels
find ./alignments -name "*.fasta" -type f | \
tview --fasta - --palette aa --columns 1-120 -o all.png
# ls with pattern
ls samples/*.bam | \
tview --bam - --ref ref.fa --region chr1:100-200 -o all_samples.png
# single file via echo
echo "my_alignment.fasta" | \
tview --fasta - --palette aa -o out.png
Python API
The core functions are available as a Python library:
from tview import fasta_panel, bam_panel, render_panels
# FASTA alignment
panel = fasta_panel("aligned.fasta", columns=list(range(1, 121)))
render_panels([panel], "output.png", palette="aa")
# BAM alignment
panel = bam_panel("sample.bam", "reference.fa", "chr1:100-200")
render_panels([panel], "output.png")
# Stack multiple panels
panels = [
bam_panel("sample1.bam", "ref.fa", "chr1:100-200"),
bam_panel("sample2.bam", "ref.fa", "chr1:100-200"),
]
render_panels(panels, "stacked.png", dpi=300, fontsize=7, cell=0.14)
# Classic (black-and-white) mode
panel = fasta_panel("aligned.fasta")
render_panels([panel], "classic.png", palette="aa", classic=True)
# Dual-reference: variant calls against SF162p3, x-axis numbered by HxB2
panel = fasta_panel(
"env_protein_aligned.fasta",
columns=list(range(1, 61)),
variant_ref="SF162p3_ref",
numbering_ref="HxB2",
max_rows=6,
)
render_panels([panel], "dual_ref.png", palette="aa", show_row_labels=True)
# Per-column tick labels
panel = fasta_panel("aligned.fasta", tick_every=1)
render_panels([panel], "per_col.png", palette="aa")
# Heterologous orange highlight (separate from dual-ref)
# Sample bases that mismatch ref_row but match secondary_ref_row render in orange + bold.
from tview.models import Panel
panel = fasta_panel("aligned.fasta")
panel = Panel(
label=panel.label,
ref_row=panel.ref_row,
seq_rows=panel.seq_rows,
total_cols=panel.total_cols,
col_labels=panel.col_labels,
secondary_ref_row=list("..."), # other strain aligned to same grid
)
render_panels([panel], "hetero.png", palette="aa")
Composing with patchworklib
draw_panels() and panel_figsize() let you draw alignments onto any
matplotlib axes, including patchworklib
Brick objects for composing multi-panel figures.
pip install patchworklib
# or
pip install tview[compose]
import patchworklib as pw
from tview import fasta_panel, draw_panels, panel_figsize
# Build alignment panel
panel = fasta_panel("aligned.fasta", columns=list(range(1, 121)))
w, h = panel_figsize([panel], fontsize=7, cell=0.14)
# Draw onto a patchworklib Brick
alignment = pw.Brick(label="alignment", figsize=(w, h))
draw_panels([panel], ax=alignment, fontsize=7, palette="aa", cell=0.14)
# Compose with other plots
scatter = pw.Brick(label="scatter", figsize=(3, 3))
scatter.scatter([1, 2, 3], [4, 5, 6])
layout = alignment / scatter # vertical stack
layout.savefig("composed.png")
This also works with standard matplotlib subplots:
import matplotlib.pyplot as plt
from tview import fasta_panel, draw_panels, panel_figsize
panel = fasta_panel("aligned.fasta")
w, h = panel_figsize([panel])
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(w + 4, max(h, 3)))
draw_panels([panel], ax1, palette="aa")
ax2.scatter([1, 2, 3], [4, 5, 6])
plt.tight_layout()
plt.savefig("side_by_side.png", dpi=300, bbox_inches="tight")
Visual Conventions
| Element | Symbol | Style |
|---|---|---|
| Match (forward) | . |
light grey |
| Match (reverse) | , |
light grey, reduced opacity |
| Mismatch | A T etc. |
colored, yellow highlight, bold |
| Mismatch (reverse) | a t etc. |
lowercase, colored, yellow highlight |
| Heterologous match | A T etc. |
orange #FF6F00, bold, yellow highlight (mismatches ref_row but matches secondary_ref_row) |
| Deletion | - |
grey dash |
| Insertion | colored bases | purple column shading |
| Gap (ref in insertion col) | - |
grey dash |
| Gap (FASTA alignment) | - |
grey dash |
Color Palettes
--palette nt (default) — Nucleotides
| Base | Color |
|---|---|
| A | green #4CAF50 |
| C | blue #2196F3 |
| G | orange #FF9800 |
| T | red #F44336 |
--palette aa — Amino Acids (Clustal-inspired)
| Group | Residues | Color |
|---|---|---|
| Hydrophobic | A V L I M F W P | blue #2196F3 |
| Positive charge | K R H | red #F44336 |
| Negative charge | D E | magenta #E040FB |
| Polar uncharged | S T N Q | green #4CAF50 |
| Special | G C Y | orange #FF9800 |
Full Argument Reference
Usage: tview [OPTIONS]
Publication-quality alignment viewer (BAM or FASTA).
Options:
--bam TEXT BAM file(s) — each becomes a panel. Use '-' for stdin.
--ref PATH Reference FASTA (required for BAM mode).
--region TEXT Genomic region chr:start-end (required for BAM mode).
--fasta TEXT Aligned FASTA file(s) — each becomes a panel. Use '-' for stdin.
--columns TEXT Column positions for FASTA, 1-based (e.g. 1-120, 5,40,690, or 5,10-20,40).
-o, --output TEXT Output image path. [default: alignment.png]
--palette [nt|aa] Color palette. [default: nt]
--dpi INTEGER Image resolution. [default: 300]
--fontsize INTEGER Base font size in points. [default: 7]
--cell FLOAT Cell size in inches. [default: 0.14]
--classic-mode Black-and-white rendering with no color highlighting.
--show Display rendered image inline via 'kitten icat' (Kitty/Ghostty).
--max-rows INTEGER Cap sample rows (FASTA) or reads (BAM) per panel.
--variant-ref TEXT FASTA header for variant calling (mismatches drawn against this).
--numbering-ref TEXT FASTA header for x-axis numbering. Renders as second top row + bottom axis when different from --variant-ref.
--show-row-labels Show sequence ID labels on the left of each row.
--tick-every INTEGER Label every Nth x-axis position. Use 1 for every column. [default: 10]
-h, --help Show this message and exit.
| Argument | Description | Default |
|---|---|---|
--bam |
BAM file(s), each becomes a panel. Use - for stdin. |
— |
--ref |
Reference FASTA (required for BAM mode) | — |
--region |
Genomic region chr:start-end (required for BAM) |
— |
--fasta |
Aligned FASTA file(s), each becomes a panel. Use - for stdin. |
— |
--columns |
Column positions for FASTA, 1-based. Supports 1-120, 5,40,690, or 5,10-20,40. |
full alignment |
-o, --output |
Output image path | alignment.png |
--palette |
Color palette: nt or aa |
nt |
--dpi |
Image resolution | 300 |
--fontsize |
Base font size in points | 7 |
--cell |
Cell size in inches (controls spacing) | 0.14 |
--classic-mode |
Black-and-white rendering with no color highlighting | False |
--show |
Display via kitten icat (Kitty/Ghostty) |
False |
--max-rows |
Cap sample rows (FASTA) or reads (BAM) per panel. In dual-ref mode, refs are always shown. | — |
--variant-ref |
(FASTA) Header name used as variant-call reference. Mismatches drawn against this row. | first sequence |
--numbering-ref |
(FASTA) Header name used for x-axis numbering. Renders as second top row + bottom axis when different from --variant-ref. |
same as --variant-ref |
--show-row-labels |
Show sequence ID labels on the left of each row. Auto-enabled when --variant-ref and --numbering-ref differ. |
False |
--tick-every |
Label every Nth x-axis position. Use 1 to label every column. |
10 |
Tips for Publication Figures
- Use
--dpi 300(default) for print,--dpi 150for drafts. - Use
--cell 0.10for denser layouts with many sequences,--cell 0.18for fewer. - Use
--fontsize 5or6when displaying wide alignments (>100 columns). - The output format is determined by the file extension:
.png,.pdf,.svgall work. - For Nature-style figures,
.pdfor.svgoutput preserves vector text. - Use
--classic-modefor textbook-style monochrome figures that reproduce well in grayscale print.
# Vector output for publication
tview \
--fasta aligned.fasta \
--palette aa \
--columns 1-120 \
--cell 0.12 \
--fontsize 6 \
-o figure_2a.pdf
FASTA Input Format
The FASTA input must be pre-aligned (e.g. by MAFFT, MUSCLE, Clustal). The first sequence is used as the reference for comparison. Gap characters (-) in the alignment are preserved and rendered as grey dashes.
>HxB2_reference
MRVK---EKYQHLWRWGWRWGTMLLGMLMICS...
>sample_001
MRVKGIRKNAQHL----WRGGTLLLGMLMICS...
>sample_002
--------------------------MLMICS...
The x-axis labels count non-gap positions in the reference sequence (1, 10, 20, ...) so position numbers always correspond to the reference residue numbering regardless of gap columns. The interval is configurable via --tick-every N.
Pass --variant-ref <name> to pick a non-first sequence as the reference for variant calling. Pass --numbering-ref <name> to layer a second reference whose non-gap positions drive an additional x-axis (typical HIV use case: HxB2 numbering with variant calls against a different strain).
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file tview-0.3.0.tar.gz.
File metadata
- Download URL: tview-0.3.0.tar.gz
- Upload date:
- Size: 2.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d77aa3b3adbcd2e90537953f6bd8c3bc934c69e344857cf13daa3618555bbc2e
|
|
| MD5 |
8c1365d04c30212bf2c06766ef18f471
|
|
| BLAKE2b-256 |
5dd5fd4d2516324c5ee697df3bf3bb77ad84cabb046b95f8a8c476e2c87a3488
|
Provenance
The following attestation bundles were made for tview-0.3.0.tar.gz:
Publisher:
publish.yml on tmsincomb/tview
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
tview-0.3.0.tar.gz -
Subject digest:
d77aa3b3adbcd2e90537953f6bd8c3bc934c69e344857cf13daa3618555bbc2e - Sigstore transparency entry: 1421166384
- Sigstore integration time:
-
Permalink:
tmsincomb/tview@d17f308d8e70fb7c94e3f6133c32d48d1fe7a1ae -
Branch / Tag:
refs/tags/v0.3.0 - Owner: https://github.com/tmsincomb
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@d17f308d8e70fb7c94e3f6133c32d48d1fe7a1ae -
Trigger Event:
push
-
Statement type:
File details
Details for the file tview-0.3.0-py3-none-any.whl.
File metadata
- Download URL: tview-0.3.0-py3-none-any.whl
- Upload date:
- Size: 24.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
06de60d173072e8e092196bdced088f72778a3495652a68b9055c9bf59dd931c
|
|
| MD5 |
858d382d8a7cc73e61e58eafaa8e6732
|
|
| BLAKE2b-256 |
a623be90d53e324e6b5eef502c51b5392b09295567572cecee3a0860bb4bcda4
|
Provenance
The following attestation bundles were made for tview-0.3.0-py3-none-any.whl:
Publisher:
publish.yml on tmsincomb/tview
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
tview-0.3.0-py3-none-any.whl -
Subject digest:
06de60d173072e8e092196bdced088f72778a3495652a68b9055c9bf59dd931c - Sigstore transparency entry: 1421166528
- Sigstore integration time:
-
Permalink:
tmsincomb/tview@d17f308d8e70fb7c94e3f6133c32d48d1fe7a1ae -
Branch / Tag:
refs/tags/v0.3.0 - Owner: https://github.com/tmsincomb
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
publish.yml@d17f308d8e70fb7c94e3f6133c32d48d1fe7a1ae -
Trigger Event:
push
-
Statement type:







