A Python package for Voronoi analysis of molecular structures

Project description

VorPy
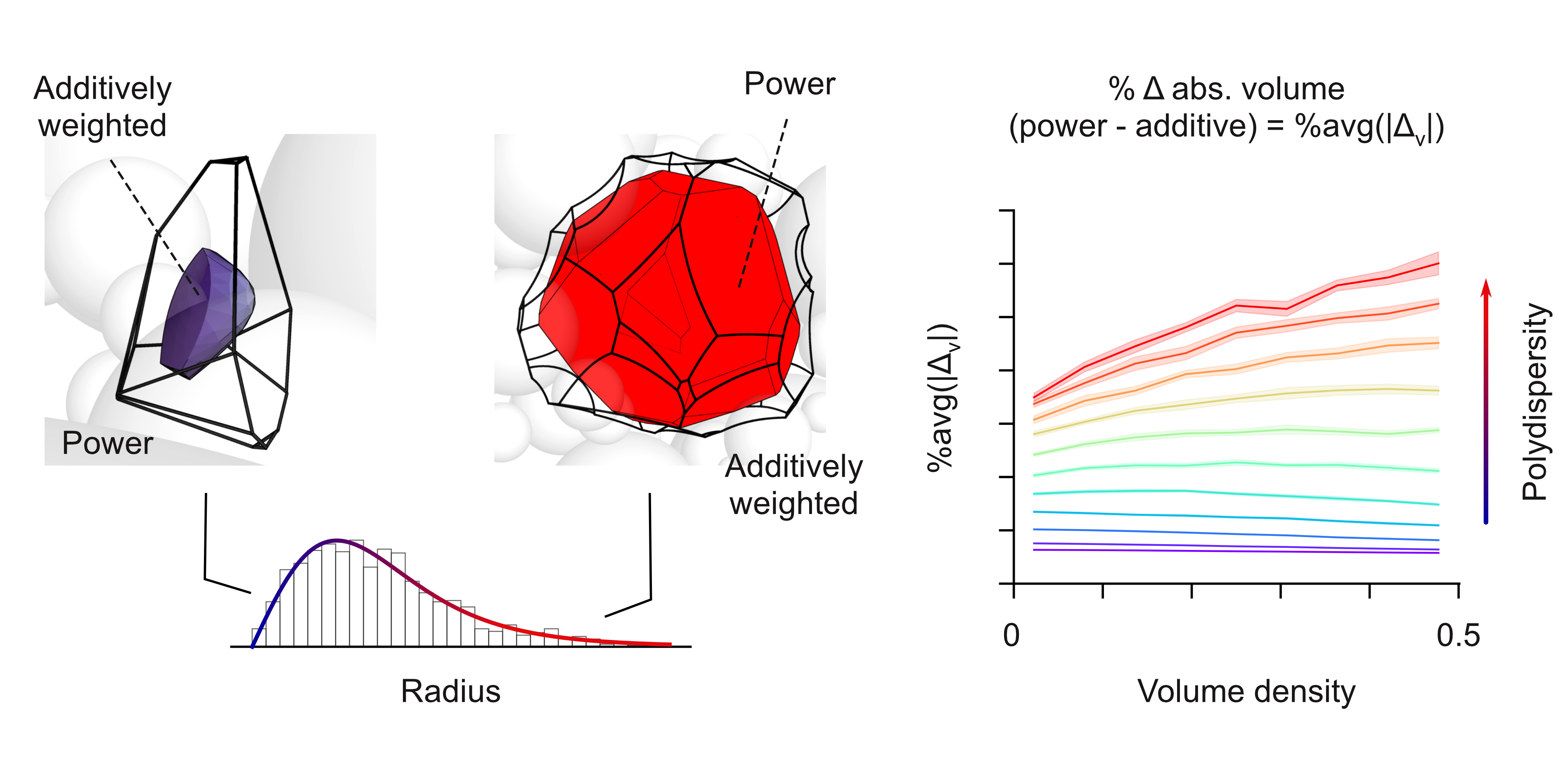
Overview
Vorpy is a spatial partitioning tool designed to solve, analyze, and export Additively Weighted, Power (Leguerre), and/or Primitive (Delaunay) Voronoi diagrams for systems of 3D spheres. This tool can accept atomic coordinate files of all major file extensions (or txt files in the right format), solve their Voronoi graphs and output visualizations and analysis of their geometry. With both a grapical user interface for simple cases and a command line interface for more complicated or cases VorPy has a robust input/output abilities.
Usage
Vorpy is available as a PyPI package. After installing it with pip (pip install vorpy3), you can launch the main GUI (see VorPy GUI) by running the following in a Python interpreter:
import vorpy as vp
vp.run()
VorPy GUI:

For detailed documentation, please visit the documentation website.
- Input File Name: The name of the ball file with the extension and the folder stripped. This will be the name of the folder that will hold the output files and will hold each of the group folders.
- Input File Information: Displays the general information about the input file and the groupings within it.
- Input Locations: Shows where the input file, the output directory, and any other loaded files can be located.
- Radii/Mass Changing: Allows the user to adjust the radii and masses of any given element and/or specific atoms in set residues (see Radii/Mass Adjustments).
- System Exports: Shows the different options for exports at the system level. Launches another window with different options including logs, ball files, set atoms (see System)
- Reset: Clears the program of any system, files, and settings that have been added.
- Groups Section: Holds each of the groups to be built and all of the corresponding selections, settings, and exports (see Groups)
- Group Name: Changes the name for the current group. Works as the name for the sub-directory for the given group's output files
- Group Selections: Allows the user to add/delete atoms/balls, residues, chains, and molecules using and index or a range. If the range entry is unfilled only the single object's index identified in the index entry box will be chosen. The indices start at 0 and correspond to the oder in which they appear in the ball file.
- Group Selection Tracker: Tracks the selections that the the user has added to the group.
- Group Build Settings: Tracks the given settings the given group will be built with
- Group Exports: Holds the different exports for the given group. There are three default options: small, medium, and large exports. If the user wants to export custom options, there is a custom exports button with all possible exports for the group.
- Run Group: Solves the group and exports the given exports. Will only solve the current group and export the current selection.
- Add/Delete Groups: Adjusts the number of groups being solved. The delete button points to the current group and will ask for permission before deleting anything.
- Help: The help button launches a window that explains all of the functions of the program and the GUI and serves as a reminder for the user (see Help Window)
- Run All Groups: Solves and exports all groups as well as the system exports. The main run function for VorPy.
Radii/Mass Adjustments:
When selected, the radii adjustment window will launch.
System Exports
The System Information frame provides an overview and management interface for your system's core configuration:
• System Name: Displays the current system's name, which is typically derived from the selected input file.
• Input File: Allows you to select the primary structure file (e.g., PDB) that defines the molecular system to be analyzed.
• Additional Files: Lists any supplementary files (such as radii, topology, or parameter files) associated with the system, if applicable.
• Output Directory: Lets you specify the folder where all generated results and exports will be saved.
To configure your system, use the "Select File" button to choose your main input file, and the "Select Directory" button to set the output location. The frame will update to reflect your selections and display relevant file information. This ensures that all subsequent operations and exports are performed using the correct files and directories.
Groups
The Groups frame provides a comprehensive interface for managing and configuring distinct groups within your system. Groups allow you to organize atoms, residues, chains, or molecules into logical sets, each of which can be processed independently with its own settings.
• Group Selection: Select specific atoms/balls, residues, chains, or molecules from the input file to define the members of each group. This enables targeted analysis or export of particular regions or components of your system.
Key features of the Groups frame include:
• Group-Specific Build Settings: Each group contains its own build settings (see the "Build Settings" tab for details), allowing you to customize parameters such as network type, maximum vertex count, box size, and surface calculation options on a per-group basis.
• Group Management: - Add or delete groups as needed to organize your workflow. - Rename groups to provide meaningful identifiers for each set. - Use the provided tabs to easily track, switch between, and manage multiple groups within your project.
• Group-Specific Export Settings: Configure export options for each group individually (see the "Export Settings" tab for more information). This includes selecting which data to export, output formats, and destination directories.
• Run Controls: - Run calculations for all groups collectively, or execute only a specific group as needed. - Each group can be processed independently, enabling flexible and efficient workflows.
• Visual Tracking: The Groups frame uses tabs or a list to help you keep track of all defined groups, their settings, and their current status.
The Groups frame is central to organizing your analysis, enabling you to tailor network construction and export options for different parts of your system. By leveraging group-specific settings, you can perform detailed, customized analyses and exports for each region of interest.
Run Function
If the package was received using PyPi, and the user wants to run multiple files, operate the package outside of the GUI, or integrate the package into an existing script, they can use the same run funciton with a file location:
import vorpy as vp
my_data = vp.run('location_to_file')
This allows the user to continue to perform operations on the data afterwords, loop through several files, change settings, change groups, or change exports without running multiple scripts.
The parameters for this operation are similar to what can be found in the GUI:
- file - The input ball/atom file holding the locations and radii of the
Command line:
If the package was downloaded over github or another repository and not prepackaged, as would be found in the The general structure of the command line follows the structure outlined below:
python vorpy/cmd.py <file>
File
- The first argument after
vorpy.pyshould be the file address of the ball or atom file. - If the file is located in the
vorpy/src/datafolder, specify the file name without the path or extension. - Accepted file extensions include
.pdb,.mol,.gro,.cif.
Load Options (Flag -l)
-l <file>
Load additional files like vertex files from previous runs, log files, Voronota vertex files, or GROMACS index files.
Setting Options (Flag -s)
-s <setting value>
Adjust various simulation parameters:
nt- Network Type: Default = Additively Weightedaw, Powerpow, Primitiveprm, or Comparecom 'type1' 'type2'mv- Maximum Vertex: Default =40bm- Box Multiplier: Default =1.25sr- Surface Resolution: Default =0.2sc- Surface Color Map: Default =viridis,plasma,rainbow, or any other matplotlib colormap (note: '_r' inverts the scheme)ss- Surface Coloring Scheme: Default = curvaturecurv, inside vs outside spheresnout, distance from centerdistsf- Surface Coloring Scale: Default = linearlin, loglog, squaredsquare, cubecubear- Adjust Radii:'element' 'value'or'atom name' 'value'or'residue' 'atom name' 'value'. To see the current values for defaults for atomic radii go to the radii file (radii.py) or enter the radii flag-r
Group Options (Flag -g)
-g <identifier>
Select specific balls or molecular elements:
b- Ball Identifier. Used with a ball index'index'or range of indices'index1'-'index2'.a- Atom Identifier. Used with an atom element'element', element name'element name', index'index', or range of indices'index1'-'index2'.r- Residue Identifier. Used with a residue name'residue name'and sequence number'sequence number'(optional), index'index', or range of indices'index1'-'index2'.c- Chain Identifier. Used with a chain name'chain name', index'index', or range of indices'index1'-'index2'.
Note: If multiple of the above components are desired in the same group use the and qualifier between components. If multiple groups are desired use multiple group flags.
Export Options (Flag -e)
-e <export_type>
Specify the intensity and type of exports:
-
Groups of Exports: Default =
large,small,medium,all -
Export choices :
Molecule File -
pdb,mol,cif,gro, Set Atoms Radii PyMol Script -set_atoms, Group Information -info, Network Logs -logs, All Surfaces in One File -surfs, All Surfaces in Separate Files -sep_surfs, All Edges in One File -edges, All Edges in Separate Files -sep_edges, All Vertices in One File -verts, All Vertices in Separate Files -sep_verts, Surrounding Surfaces -shell, Surrounding Edges -shell_edges, Surrounding Vertices -shell_verts, Group Atoms -atoms, Atoms Surrounding Group -surr_atoms
Command Line Notes
- Each option flag and its arguments must be separated by spaces.
- To use multiple commands for a single option use 'and' or repeat the flag (except for groups to avoid creating multiple groups).
- Any range can be set with a hyphen and no space (e.g.
-g a 0-100is a group of the first 101 atoms)
Requirements and Dependencies
Requirements
- Python 3.8+
vorPy is developed and tested with Python 3.8 and above.
Python Dependencies
The following Python packages are required to run vorPy:
numpyscipymatplotlibpandasPillowtkinter(for GUI)pytest(for running tests, optional)
You can install all dependencies using:
pip install -r requirements.txt
Output Examples
The outputs for the program are either informative or visualizations of the data. In th
Examples
Example 1: Simple Molecular Visualization In this example, we look at the molecule EDTA which binds to harmful magnesium ions in food to neutralize them. The EDTA/Mg complex is preloaded into vorPy and can be run simply with the following command:
python vorpy.py EDTA_Mg
This will solve the additively weighted Voronoi diagram for the EDTA molecule and the Mg atom. Once solved, the program will output the default outputs (see outputs).
Example 2: Separately solve the tyrosine 2 and methionine 1 residues of the cambrin molecule, calculate their interface, and export the large export type of the results
python vorpy.py cambrin -s sr 0.05 and mv 80 -g tyr 2 -g met 1 -c iface -e large
Example 3: Calculate the primitive and power networks for the mg atom in the EDTA_Mg molecule and compare the difference
python vorpy.py EDTA_Mg -s nt compare prm pow -g mg
Example 4: Solve the network for hairpin and export the shell with the inside and outside parts of the surfaces highlighted at a high resolution
python vorpy.py hairpin -s ss nout and sr 0.01 -e shell and pdb
License
This project is licensed under the MIT License - see the LICENSE file for details.
Citation
If you use VORPY in your research, please cite:
@software{vorpy2024,
author = {John Ericson},
title = {VORPY: A Python package for Voronoi analysis of molecular structures},
year = {2024},
publisher = {GitHub},
url = {https://github.com/jackericson98/vorpy}
}
Contact
- Email: jericson1@gsu.edu
- Site: ericsonlabs.com
- Phone: +1 (404)-413-5491
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file vorpy3-3.1.1.tar.gz.
File metadata
- Download URL: vorpy3-3.1.1.tar.gz
- Upload date:
- Size: 40.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
891801948ef70b57b7fb1c82b446a041b834d1300419d129338b627aef23ac64
|
|
| MD5 |
c65c94126ef14d0721d52bba0111d284
|
|
| BLAKE2b-256 |
f445e9f33d4f14a02a199d03e1bbc5faea936c634d2a05ff0fe627381b01b0f3
|
File details
Details for the file vorpy3-3.1.1-py3-none-any.whl.
File metadata
- Download URL: vorpy3-3.1.1-py3-none-any.whl
- Upload date:
- Size: 40.9 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
afa61c1b4a7058cf88c91aa716386571e66ab6457c0abaef26a512e62dea076d
|
|
| MD5 |
c8bb7033df662ddaa4f79b852fd83d82
|
|
| BLAKE2b-256 |
a247641eb244700cec54184063dc35f175b69a2388889fb31974474a3ba7bb83
|







