Comprehensive genotyping tool for targeted long-read sequencing analysis

Project description









DAJIN2 is a genotyping tool for genome-edited samples using nanopore-targeted sequencing.
DAJIN2 takes its name from the Japanese phrase 一網打尽 (Ichimou DAJIN in Japanese; Yīwǎng Dǎjìn in Chinese),
meaning “to capture everything in a single sweep.”
This reflects the tool’s design philosophy: comprehensive detection of both intended and unintended genome editing outcomes in one analysis.
🌟 Features
-
Comprehensive Mutation Detection
DAJIN2 can detect a wide range of genome editing events in nanopore-targeted regions, from point mutations to structural variants.
It is particularly effective at identifying unexpected mutations and complex mutations, such as insertions within deleted regions. -
Highly Sensitive Allele Classification
Supports classification of mosaic alleles, capable of detecting minor alleles present at approximately 1%. -
Intuitive Visualization
Genome editing results are visualized in an intuitive manner, enabling rapid and easy identification of mutations. -
Multi-Sample Support
Batch processing of multiple samples is supported, allowing efficient execution of large-scale experiments and comparative studies. -
Simple Installation and Operation
Requires no specialized computing environment and runs smoothly on a standard laptop.
Easily installable via Bioconda or PyPI, and usable via the command line.
🛠 Installation
System Requirements
Hardware
- Runs on a standard laptop
- Recommended memory: 16 GB or more
[!NOTE] DAJIN2 is the successor to DAJIN, which required a GPU for efficient computation due to its use of deep learning.
In contrast, DAJIN2 does not use deep learning and does not require a GPU.
Therefore, it runs smoothly on typical laptops.
Software
- Python 3.10-3.12
- Unix-based environment (Linux, macOS, WSL2, etc.)
[!IMPORTANT] For Windows Users
DAJIN2 is designed to run in a Linux environment.
If you are using Windows, please use WSL2 (Windows Subsystem for Linux 2).
From Bioconda (Recommended)
conda create -y -n env-dajin2 -c conda-forge -c bioconda DAJIN2
conda activate env-dajin2
From PyPI
pip install DAJIN2
[!IMPORTANT] DAJIN2 is actively being developed and improved.
Please make sure you are using the latest version to take advantage of the newest features.1. To check your current version
DAJIN2 --version2. Check the latest version
https://github.com/akikuno/DAJIN2/releases3. Reinstall DAJIN2 if the installed version is not the latest
conda deactivate conda remove -y -n env-dajin2 --all conda create -y -n env-dajin2 -c conda-forge -c bioconda DAJIN2 conda activate env-dajin2or
pip install -U DAJIN2
[!CAUTION] If you encounter any issues during the installation, please refer to the Troubleshooting Guide
💻 Usage
Required Files
1. FASTQ/FASTA/BAM Files for Sample and Control
In DAJIN2, a control that has not undergone genome editing is necessary to detect genome-editing-specific mutations. Specify a directory containing the FASTQ/FASTA (both gzip compressed and uncompressed) or BAM files of the genome editing sample and control.
Basecalling with Dorado
- For basecalling with Dorado (
dorado basecalleranddorado demux), the following file structure will be output:
bam_pass
├── barcode01
│ └── EXP-PBC096_barcode01.bam
├── barcode02
│ └── EXP-PBC096_barcode02.bam
├── ...
└── unclassified
│ └── EXP-PBC096_unclassified.bam
[!IMPORTANT] Store each BAM file in a separate directory.
The directory names can be set arbitrarily.
[!TIP] For detailed Dorado usage, see DORADO_HANDLING.md.
- Similarly, store the FASTA files outputted after sequence error correction with
dorado correctin separate directories.
dorado_correct
├── barcode01
│ └── EXP-PBC096_barcode01.fasta
└── barcode02
└── EXP-PBC096_barcode02.fasta
[!NOTE] Correction with
dorado correctmay mask minor editing outcomes that can arise from genome editing.
Therefore, unlessdorado correctis required, we recommend using the BAM files output bydorado basecalleranddorado demux.
Basecalling with Guppy
After basecalling with Guppy, the following file structure will be output:
fastq_pass
├── barcode01
│ ├── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_0_0.fastq.gz
│ ├── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_10_0.fastq.gz
│ └── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_11_0.fastq.gz
└── barcode02
├── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_0_0.fastq.gz
├── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_10_0.fastq.gz
└── fastq_runid_b347657c88dced2d15bf90ee6a1112a3ae91c1af_11_0.fastq.gz
[!CAUTION] Although DAJIN2 can process Guppy-generated data, Guppy is no longer supported by Oxford Nanopore Technologies.
Please use Dorado for basecalling and demultiplexing.
2. FASTA File Including Anticipated Allele Sequences
The FASTA file should contain descriptions of the alleles anticipated as a result of genome editing.
[!IMPORTANT] A header name
>controland its sequence are necessary.
If there are anticipated alleles (e.g., knock-ins or knock-outs), include their sequences in the FASTA file too. These anticipated alleles can be named arbitrarily.
Below is an example of a FASTA file:
>control
ACGTACGTACGTACGT
>knock-in
ACGTACGTCCCCACGTACGT
>knock-out
ACGTACGT
Here, >control represents the sequence of the control allele, while >knock-in and >knock-out represent the sequences of the anticipated knock-in and knock-out alleles, respectively.
[!IMPORTANT] Ensure that both ends of the FASTA sequence match those of the amplicon sequence.
If the FASTA sequence is longer or shorter than the amplicon, the difference may be recognized as an indel.
Using BED Files for Genomic Coordinates
[!NOTE] BED file is an optional.
If the reference genome is not from UCSC, or if the external servers that DAJIN2 depends on (UCSC Genome Browser and GGGenome) are unavailable, you can specify a BED file using the -b/--bed option to run offline.
[!TIP] Access to the UCSC Genome Browser or GGGenome servers may occasionally be unavailable. Therefore, we generally recommend using
-b/--bedinstead of--genome.
When using the -b/--bed option with a BED file, please ensure:
Use BED6 format (6 columns required):
chr1 1000000 1001000 mm39 195154279 +
Column descriptions:
- Column 1: Chromosome name (e.g., chr1, chr2)
- Column 2: Start position (0-indexed)
- Column 3: End position (0-indexed)
- Column 4: Name (genome ID)
- Column 5: Score (chromosome size for proper IGV visualization)
- Column 6: Strand (+ or -, must match FASTA allele orientation)
[!NOTE]
For the score field (column 5), please enter the size of the chromosome specified in column 1.
While the original BED format limits scores to 1000, DAJIN2 accepts chromosome sizes without any issue.
[!NOTE] Chromosome sizes can be found at:
https://hgdownload.soe.ucsc.edu/goldenPath/[genome]/bigZips/[genome].chrom.sizes
(e.g., https://hgdownload.soe.ucsc.edu/goldenPath/mm39/bigZips/mm39.chrom.sizes)
[!IMPORTANT]
Strand orientation must match. The strand field (column 6:+or-) in your BED file must match the strand orientation of your FASTA allele sequences.
- If your FASTA allele sequence is on the forward strand (5' to 3'), use
+in the BED file- If your FASTA allele sequence is on the reverse strand (3' to 5'), use
-in the BED file
[!TIP] For detailed BED file usage, see BED_COORDINATE_USAGE.md.
Single Sample Analysis
DAJIN2 supports single-sample analysis (one sample vs one control).
DAJIN2 <-c|--control> <-s|--sample> <-a|--allele> <-n|--name> \
[-g|--genome] [-b|--bed] [-t|--threads] [--no-filter] [-h|--help] [-v|--version]
Options:
-c, --control Specify the path to the directory containing a control FASTQ/FASTA/BAM file.
-s, --sample Specify the path to the directory containing a sample FASTQ/FASTA/BAM file.
-a, --allele Specify the path to the FASTA file.
-n, --name (Optional) Set the output directory name. Default: 'Results'.
-b, --bed (Optional) Specify the path to BED6 file containing genomic coordinates. Default: '' (empty string).
-g, --genome (Optional) Specify the reference UCSC genome ID (e.g., hg38, mm39). Default: '' (empty string).
-t, --threads (Optional) Set the number of threads. Default: 1.
--no-filter (Optional) Disable minor allele filtering (keep alleles below 0.5%). Default: False.
-h, --help Display this help message and exit.
-v, --version Display the version number and exit.
Example
# Download the example dataset
curl -LJO https://github.com/akikuno/DAJIN2/raw/main/examples/example_single.tar.gz
tar -xf example_single.tar.gz
# Run DAJIN2
DAJIN2 \
--control example_single/control \
--sample example_single/sample \
--allele example_single/stx2_deletion.fa \
--name stx2_deletion \
--bed example_single/stx2_deletion.bed \
--threads 4
Rare Mutation Detection with --no-filter
By default, DAJIN2 filters out alleles with read counts below 0.5% (5 reads out of 100,000 downsampled reads) to reduce noise and improve accuracy. However, when analyzing rare mutations or somatic mosaicism where minor alleles may be present at very low frequencies, you can use the --no-filter option to disable this filtering.
When to use --no-filter:
- Detecting rare somatic mutations (< 0.5% frequency)
- Analyzing samples with suspected low-level mosaicism
- Research requiring detection of all possible alleles regardless of frequency
Usage:
DAJIN2 \
--control example_single/control \
--sample example_single/sample \
--allele example_single/stx2_deletion.fa \
--name stx2_deletion \
--bed example_single/stx2_deletion.bed \
--threads 4 \
--no-filter
[!CAUTION] Using
--no-filtermay increase noise and false positives in the results. It is recommended to validate rare alleles through additional experimental methods.
Batch Processing
By using the batch subcommand, you can process multiple samples simultaneously.
For this purpose, a CSV or Excel file consolidating the sample information is required.
[!NOTE] For guidance on how to compile sample information, please refer to this document.
Required columns: sample, control, allele, name
Optional columns: genome, bed (or genome_coordinate), and any custom columns
Example CSV with BED files:
sample,control,allele,name,bed
/path/to/sample1,/path/to/control1,/path/to/allele1.fa,experiment1,/path/to/coords1.bed
/path/to/sample2,/path/to/control2,/path/to/allele2.fa,experiment2,/path/to/coords2.bed
[!TIP] We recommend using the same name in the
namecolumn for sample sets you want to compare.
Sample sets with the same value in thenamecolumn are treated as a single group, and their outputs can be consolidated into one.
Here is an example 👉 batch.csv
DAJIN2 batch <-f|--file> [-t|--threads] [--no-filter] [-h]
Options:
-f, --file Specify the path to the CSV or Excel file.
-t, --threads (Optional) Set the number of threads. Default: 1.
--no-filter (Optional) Disable minor allele filtering (keep alleles below 0.5%). Default: False.
-h, --help Display this help message and exit.
Example
# Download the example dataset
curl -LJO https://github.com/akikuno/DAJIN2/raw/main/examples/example_batch.tar.gz
tar -xf example_batch.tar.gz
# Run DAJIN2 batch
DAJIN2 batch --file example_batch/batch.csv --threads 4
GUI (Graphical User Interface) Mode
DAJIN2 can launch a web app with the following command:
DAJIN2 gui
When executed, your default web browser will open and display the following GUI at http://localhost:{PORT}/.
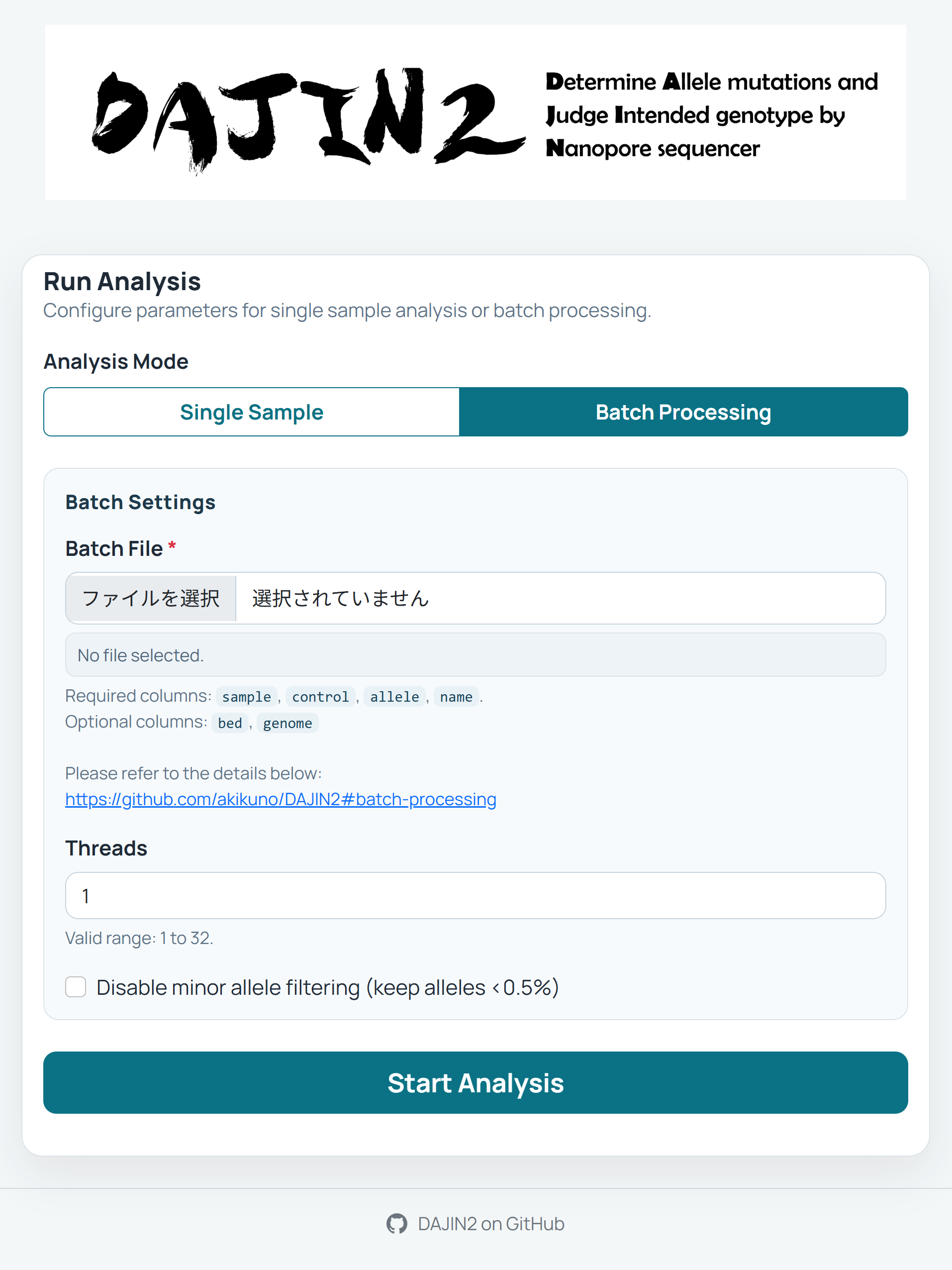
[!NOTE] If the browser does not launch automatically, please open your browser manually and navigate to
http://localhost:{PORT}/.
Single Sample Analysis via GUI
-
Project Setup
- Project Name: Enter any analysis name
- Directory Upload: Select directories containing sample or control FASTQ/FASTA/BAM files
- Allele FASTA: Upload FASTA file containing expected allele sequences
- BED File (optional): Upload BED6 format file to specify genomic coordinates
- Reference Genome (optional): Specify UCSC genome ID (e.g.,
hg38,mm39)
-
Parameter Configuration
- Threads: Set the number of CPU threads to use
- No Filter: Enable to detect rare mutations below 0.5% frequency
-
Run Analysis
Click "Start Analysis" and progress will be displayed in real time. -
View Results
After completion, the output folder path will be displayed and result files will be accessible.
Batch Processing via GUI
-
Prepare Batch File
Create a CSV or Excel file with columns:sample,control,allele,name. -
Upload Batch File
Use the "Batch Processing" tab to upload your configuration file. -
Parameter Configuration
- Threads: Set the number of CPU threads to use
- No Filter: Enable to detect rare mutations below 0.5% frequency
-
Run Analysis
Click "Start Analysis" and progress will be displayed in real time. -
View Results
After completion, the output folder path will be displayed and result files will be accessible.
📈 Reports
Upon completion of DAJIN2 processing, a directory named DAJIN_Results/{NAME} is generated.
Inside the DAJIN_Results/{NAME} directory, the following files can be found:
DAJIN_Results/tyr-substitution
├── BAM
│ ├── control
│ ├── tyr_c230gt_01
│ ├── tyr_c230gt_10
│ └── tyr_c230gt_50
├── DAJIN2_log_20260127_140954_076887.txt
├── FASTA
│ ├── tyr_c230gt_01
│ ├── tyr_c230gt_10
│ └── tyr_c230gt_50
├── HTML
│ ├── tyr_c230gt_01
│ ├── tyr_c230gt_10
│ └── tyr_c230gt_50
├── MUTATION_INFO
│ ├── tyr_c230gt_01.csv
│ ├── tyr_c230gt_10.csv
│ └── tyr_c230gt_50.csv
├── VCF
│ ├── tyr_c230gt_01
│ ├── tyr_c230gt_10
│ └── tyr_c230gt_50
├── launch_report_mac.command
├── launch_report_windows.bat
└── read_summary.xlsx
1. launch_report_windows.bat / launch_report_mac.command
On Windows, double-click launch_report_windows.bat.
On macOS, double-click launch_report_mac.command.
Your browser will open and display the report.

Demo video:
https://github.com/user-attachments/assets/e2de7b56-94c8-4361-a9d3-54c30d53720c
[!TIP] Clicking on an allele of interest in the stacked bar chart allows you to view detailed information on the mutation (right panel above on figure, and video).
In the report, Allele type indicates the allele category, and Percent of reads shows the proportion of reads.
Allele type categories:
- {Allele name}: Alleles that perfectly match a user-defined allele in the FASTA file
- {Allele name} with indels: Alleles similar to a user-defined allele but with a few-base substitution, deletion, insertion, or inversion
- unassigned insertion/deletion/inversion: Alleles with deletions, insertions, or inversions of 10 bases or more that are not defined by the user
[!WARNING]
In PCR amplicon sequencing, Percent of reads may not match the true allele proportions due to amplification bias.
This effect can be pronounced when large deletions are present, potentially distorting the actual allele ratios.
2. read_summary.xlsx
read_summary.xlsx lists the read counts and proportions for each allele.
The stacked bar chart in the report is a visualization of read_summary.xlsx.
Use it as reference when preparing figures for publications.
3. BAM and VCF
The BAM and VCF directories contain BAM and VCF files classified by allele.
[!NOTE]
If--bedor--genomeis not specified, reads are aligned to the control allele in the input FASTA file.
4. FASTA and HTML
The FASTA directory stores FASTA files for each allele.
The HTML directory stores per-allele HTML files with color-highlighted mutations.
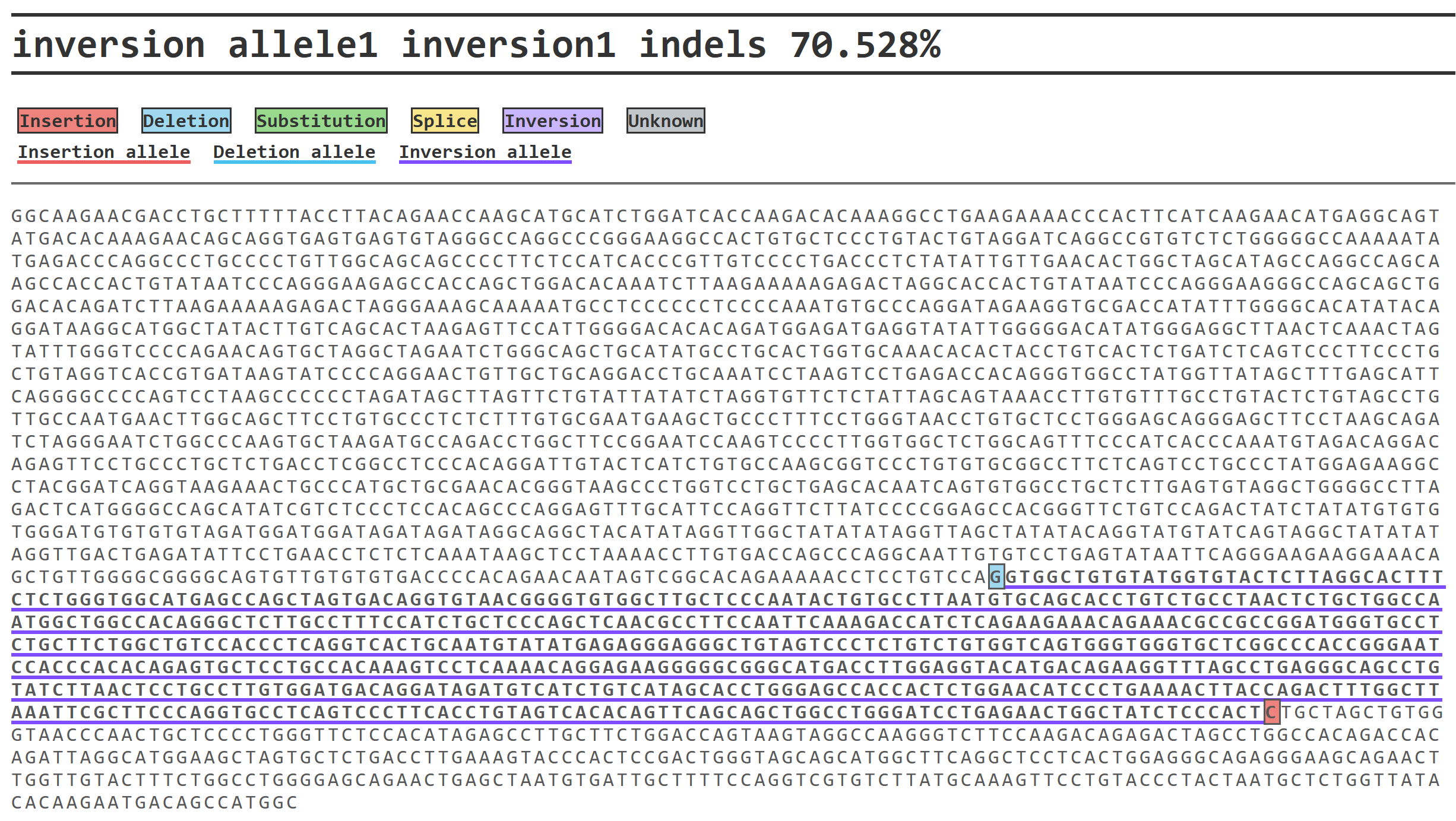
An example of a Tyr point mutation (green) is shown below:

DAJIN2 also extracts representative SV alleles (Insertion, Deletion, Inversion) in the sample and underlines SV regions.
Below is an example where a deletion (light blue) and an insertion (red) are observed at both ends of an inversion (purple underline).
📣 Feedback and Support
We welcome your questions, bug reports, and feedback.
Please use the following Google Form to submit your report:
👉 Google Form
If you have a GitHub account, you can also submit reports via
👉 GitHub Issues
Please refer to CONTRIBUTING for how to contribute and how to verify your contributions.
[!NOTE] For frequently asked questions, please refer to this page.
🤝 Code of Conduct
Please note that this project is released with a Contributor Code of Conduct.
By participating in this project you agree to abide by its terms.
📄 References
For more information, please refer to the following publication:
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file dajin2-0.9.3.tar.gz.
File metadata
- Download URL: dajin2-0.9.3.tar.gz
- Upload date:
- Size: 1.2 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
79e50b1b544b3e67f4d1f0dc04b2ff7b218574151fd3c93f4c4c4dfe9bae864d
|
|
| MD5 |
b73b51971e61e6ab38946af9251496fd
|
|
| BLAKE2b-256 |
c5ffb6b833fe640c963c9c36471005991c7b42b6adae7aa5313c7f2e69850eb7
|
File details
Details for the file dajin2-0.9.3-py3-none-any.whl.
File metadata
- Download URL: dajin2-0.9.3-py3-none-any.whl
- Upload date:
- Size: 1.2 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
032f9d333223923867430dbb88d719b8d5b85beda6cac44e39157a6abb8c89c3
|
|
| MD5 |
513a1097833bb4e974f84a6bc607d453
|
|
| BLAKE2b-256 |
858e3d0c2a467339720cb3b3a30b758cbde8232e09aa18b8a3324a47f2b0fc86
|







